6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffe4e82c0000007506000001000400
6go6ckt5b5idvals|983
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Complete congenital arhinia is an extremely rare embryologic disorder with less than 50 reported cases [
1]. The disorder is usually associated with other craniofacial abnormalities such as cleft lip, palate, underdeveloped maxilla and rudimentary nasal bones [
2-
7]. Associated conditions that play a part and impact the patient’s prognosis include microphthalmia, coloboma, hypertelorism, absent nasolacrimal ducts [
8], Treacher-Collins syndrome [
9] and a plethora of other central nervous system defects [
6]. The pathogenesis of complete congenital arhinia although poorly understood, involves various disturbances in the pathway of embryologic nasal development. Various mechanisms regarding the embryologic disturbance include reduced growth of median and lateral nasal processes [
10], defective invagination of the nasal placodes, premature fusion of the median nasal processes, and abnormal migration of neural crest cells and failure of resorption of the nasal epithelial plugs.
Case Report
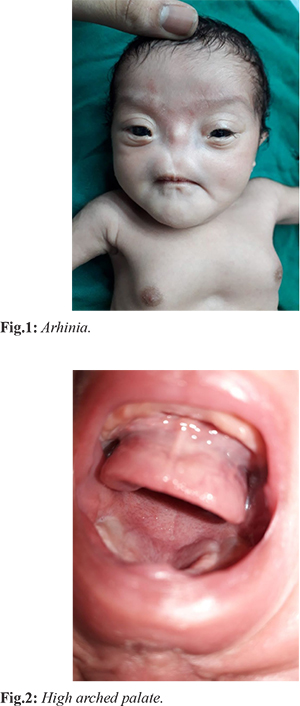
Our case was a full-term girl born by normal delivery out of a non-consanguineous marriage. The 25-year-old primigravida mother reported a normal pregnancy, with no history of any amniotic fluid imbalance or drug intake during pregnancy. Since she hailed from a rural area there was little or no contact with a gynaecologist. Just after delivery the local doctor at Bhandara noticed an absent nose with the root of the nose also being absent [Fig.1]. The neonate also had a high-arched palate [Fig.2]. The baby was breathing with her mouth and had respiratory distress. She was then referred from the rural hospital to Government Medical College, Nagpur which is a tertiary care referral centre for expert management. On admission, no other physical abnormality was seen. Routine laboratory findings were also within normal limits.
References were sent to various departments including plastic surgery, ophthalmology, cardiac surgery, otorhinolaryngology and a multi-disciplinary approach was followed. Co-existing findings included a bilateral infero-nasal typical iris coloboma, telecanthus, mongoloid slant. The child’s retina and nasolacrimal duct could not be evaluated as it needed a general anaesthesia back-up which was contraindicated due to absent nasal openings. A spiral axial CT scan with 3 mm sections was obtained which showed features of a hypoplastic nose, nasal septum and bilateral ethmoidal sinuses along with narrowing of bilateral choanae [Fig. 3-7]. Both nostrils and nasal cavity appeared rudimentary. There were also features suggestive of a hypoxic ischaemic encephalopathy. MRI of the brain showed a normal brain parenchyma with non-visualization of the nose and bilateral nasal cavities.
We advised the child to regularly follow up with us, with a plan to first operate the patient at 6 months of age to create a nasal airway with skin grafting followed by nasal splints followed by a reconstruction of the external nose at a later date once the child’s nasal and maxillofacial development is complete. To assist with pre-surgical planning and surgical stent manufacture in case this patient’s nasolacrimal ducts have to be created, accurate three-dimensional models will be needed. We plan to do this by manufacturing a physical prototype of the skull using 3D computer aided design (CAD) data.
Discussion
The embryologic development of the face and the nose starts at 3 weeks of gestation and continues till the 10th week [
1]. Two ridge shaped structures known as nasal placodes which ultimately form the lateral and medial nasal swellings develop on the 4th week of gestation. These placodes invaginate on the underlying mesenchymal tissue to form nasal pits around the 5th week of gestation and the medial nasal swellings fuse to form the nasal septum. The 6th week involves the maxillary and the frontal processes fusing to form the primitive palatal shell. The cells from the nasal pit migrate posteriorly to form the primitive nasal cavity. Fusion or merging of all the above processes occurs by the 7th to 8th week of gestation. There are a few postulations of how an arhinia may develop which include failure of the medial and lateral nasal processes to grow, premature fusion of the medial nasal processes, and lack of resorption of the nasal epithelial plugs.
From literature review, congenital arhinia does not seem to be associated with any specific genetic mutation. Most of these patients have normal chromosomal analysis which is under process in our patient, except for a few with abnormal karyotyping. Antenatal ultrasound can be useful in detecting congenital arhinia; most of the patients with this anomaly being diagnosed at birth. Early diagnosis is always preferable for better education of the parents and allows the medical team to be better prepared for delivery and its complications. Initial management usually involves securing and protecting the airway through endotracheal intubation at the time of birth. Placement of the endotracheal tube is not ideal for long-term management of these patients as it prevents the infant from feeding orally [
11]. A tracheostomy tube should be placed once the patient is stable and fit for surgery which will facilitate oral feeding and contribute to the neonate’s overall health. The tube should be removed once the patient is able to maintain saturation through mouth breathing while sleeping as well as feeding. There is a multitude of dilemmas as regards the reconstruction of these patients. The various degrees of nasal deformity or complete absence require each case to be studied carefully to reach an individualized surgical plan. The reconstruction cannot be done without a multi-disciplinary approach. The arhinia reconstruction involves a two-pronged approach: reconstruction of the nasal cavity and reconstruction of the external nose. The two methods of nasal cavity reconstruction include creation of two separate nasal cavities and then maintaining their patency with silicon tubes and the second method involves a Le Fort II maxillary osteotomy [
1]. The staged reconstruction is more common rather than simultaneous reconstruction [
1,
12-
14]. The age at which the patient can undergo such surgery is debatable. Some surgeons prefer waiting until the patient reaches adolescence whilst, others prefer intervening early to lessen the psychological impact on the patient [
15]. Our literature search showed that a patient with arhinia can have successful facial reconstructive surgery and later lead a normal life after surgery. The debate for the time of reconstruction in these cases is unending and leaning towards staged reconstruction. The importance of a multi-disciplinary approach in the management cannot be stressed enough. In the immediate future, the care-giver of such patients will have to be taught to master the skill of caring for a tracheostomy and feeding via an orogastric tube.
Conclusion
Congenital arhinia is rarely seen in everyday practice. Self-image and self-perception of a person’s deformities have varied impacts on the patient’s acceptance of themselves, surroundings and their emotional and social well-being. These patients will rarely experience a life with a normal facial appearance which may have social and psychological implications.
Contributors: SSK: manuscript writing, literature review and data acquisition; SBP: manuscript editing, and literature review; AMW: literature review and discussion; SVP: critical inputs into the manuscript. SSK will act as a study guarantor. All authors approved the final version of this manuscript and are responsible for all aspects of the study.
Funding: None; Competing interests: None stated.
References
- Zhang MM, Hu YH, He W, Hu KK. Congenital arhinia: a rare case. Am J Case Rep. 2014;15:115-118.
- Majewski S, Donnenfeld AE, Kuhlman K, Patel A. Second-trimester prenatal diagnosis of total arhinia. J Ultrasound Med. 2007;26:391-395.
- Tessier P, Ciminello FS, Wolfe A. The arhinias. Scand J Plast Reconstr Surg Hand Surg. 2009;43:177-196.
- Albernaz V, Castillo M, Mukherji SK, Ihmeidan IH. Congenital arhinia. Am J Neuroradiol. 1996;17:1312-1314.
- Cusick W, Sullivan CA, Rojas B, Poole AE, Poole DA. Prenatal diagnosis of total arhinia. Ultrasound Obstet Gynecol. 2000;15:259-261.
- Olsen O, Gjelland K, Reigstad H, Rosendahl K. Congenital absence of the nose: a case report and literature review. Pediatr Radiol. 2001;31:225-232.
- Brusati R, Colletti G. The role of maxillary osteotomy in the treatment of arhinia. J Oral Maxillofac Surg. 2012;70:361-368.
- Weinberg A, Neuman A, Benmeir P, Lusthaus S, Wexler MR. A rare case of arhinia with severe airway obstruction: a case report and review of the literature. Plast Reconstr Surg. 1993;91:146-149.
- Hansen M, Lucarelli MJ, Whiteman DA, Mulliken JB. Treacher Collins syndrome: phenotype variability in a family including an infant with arhynia and uveal colobomas. Am J Med Genet. 1996;61:71-74.
- Tanyeri B, Aygun C, Ceyhan M, Guneren E. Congenital arhinia. Int J Pediatr Otorhinolaryngol Extra. 2007;2:158-160.
- Gallacher D, Hart K, Kotecha S. Common respiratory conditions of the newborn. Breathe. 2016;12:30-42.
- Lutolf U. Bilateral aplasia of the nose: a case report. J Maxillofac Surg. 1976;4:245-249.
- LaTrenta GS, Choi HW, Ward RF, Hoffman L, Neidich JA. Complete nasal agenesis with bilateral microphthalmia and unilateral duplication of the thumb. Plast Reconstr Surg. 1995;95:11011104.
- Brusati R, Colletti G. The role of maxillary osteotomy in the treatment of arhinia. J Oral Maxillofac Surg. 2012;70:361-368.
- Fischer H, Gubisch W, Jurkovic D. Nasal reconstruction in children. Facial Plastic Surgery. 2014;30:357-364.