6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff24fc2e0000006407000001000c00
6go6ckt5b5idvals|2011
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Erdheim-Chester Disease (ECD) is a rare disease characterized by multi-organ histiocytic infiltration by CD68 positive, CD1a negative foamy histiocytes [
1,
2]. The exact prevalence is unknown, however approximately 550 cases have been described in the literature. Recent detection of a mutation in the BRAF proto-oncogene has been identified in more than half of ECD patients, which has suggested ECD is a clonal disorder and has offered further insight to diagnosis and management options [
3]. It can present with a conundrum of perplexing clinical symptoms and therefore can be difficult to diagnose if unaware of the disease. The most common sites involved include lungs, osseous structures, peri-nephric involvement, skin, and the CNS. Our case is a rare manifestation of multiple organ involvement, which can all be explained by ECD.
Case Report
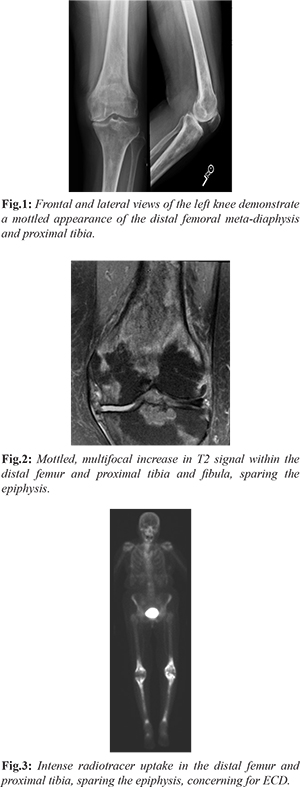
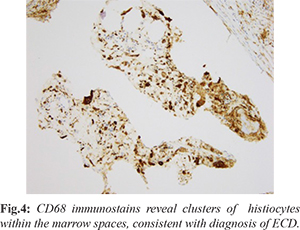
Our patient is a 75-year-old with past medical history of breast cancer status post-lumpectomy, lung cancer status post left lower lobectomy, now presenting with 5 months of left knee pain that started after gardening. Of note, she had question of fibrosing non-specific interstitial pneumonitis (NSIP) characterized on multiple chest CT scans, obtained for surveillance for history of lung cancer. Initial work up of her left knee pain included a radiograph which demonstrated a diffusely mottled appearance of the distal femoral meta-diaphysis and proximal tibia with ill-defined areas of sclerosis [Fig.1], for which an MRI was recommended for further evaluation. The initial differential diagnosis included metabolic diseases, including Paget’s, metastasis given her cancer history, and other less common entities. Further evaluation with MRI was performed, which revealed multiple areas of signal abnormalities involving the distal femur and proximal tibia, while sparing the epiphysis [Fig.2]. A bone biopsy was performed which revealed marrow space fibrosis without a specific diagnosis. Because the patient continued to have pain, a bone scan was performed, which revealed intense radiotracer uptake in the distal femur and proximal tibia bilaterally [Fig.3], while sparing the epiphysis, at which point the diagnosis of Erdheim-Chester disease was questioned. Pathology was asked to reexamine the biopsy specimens and confirmation of the diagnosis of ECD was done. A CD68 stain was performed, which is a histiocytic marker, revealing multiple clusters of histiocytes within the marrow spaces, favoring the diagnosis of ECD [Fig.4].
Reevaluation of the chest CT scan performed for surveillance of prior lung cancer demonstrated smooth progressive peripheral septal thickening, without honey combing with preserved lung volumes [Fig.5]. While these lung findings were initially attributed to NSIP, they are pathognomic of lung involvement by ECD. MRCP performed for surveillance of intraductal papillary mucinous neoplasm also demonstrated decreased T2 signal in the bilateral collecting systems, likely due to histiocytic infiltration from ECD, which typically infiltrates the kidneys and collecting systems [Fig.6].
Discussion
ECD is a rare disease which can manifest in different clinical scenarios, with common sites of involvement including the skeleton, CNS, cardiovascular system, lungs, retro-peritoneum, peri-renal spaces, and skin. Our case is a rare presentation of a patient who had lung, skeletal, and renal involvement. It is characterized by multi-organ infiltration by non-Langerhans cell histiocytes, which lack Birbeck granules and S-11 proteins. There is no known familial or genetic predisposition, however recently, a mutation of the proto-oncogene BRAF (BRAFV600E) has been found in over half of the cases.
The most frequent symptom is bone pain, particularly of the knees and ankles [
4,
5]. Although our case showed typical manifestations of ECD by chest CT, the diagnosis was not made until the patient presented with knee pain. The best diagnostic clue is a triad of osteoslerotic bone lesions, peri-renal soft tissue, also known as a “hairy kidney” appearance, and pleural or diffuse septal thickening. Additional common symptoms include bilateral exophthalmos from infiltration of the retrobulbar fat and diabetes insipidus [
3,
5].
Long bone osteosclerosis of the metaphysis and diaphysis is the most characteristic finding. In particular, the lower extremities are more commonly involved than the upper extremities. The typical nuclear medicine findings include abnormally increased uptake of technetium-99m on bone scan that is bilateral and symmetric and spares the epiphysis. This was the finding that led to the diagnosis in our case. Lytic bone lesions have been described but are less common.
Pulmonary infiltration is less common, however when present, manifests as diffuse interlobular smooth septal thickening, which is also secondary to infiltration by histiocytes. This may result in dyspnea in many cases. This was present in our patient. Centrilobular nodular opacities and diffuse ground glass attenuation are also common. Although not the most common organ involved, the most common cause of death in ECD is pulmonary fibrosis and respiratory distress.
Retro-peritoneal infiltration is common however our patient did not have this finding. This usually is asymptomatic and may sometimes present with lower back pain. CNS involvement may include intracerebral or intramedullary lesions and infiltration of the pituitary stalk may cause diabetes insipidus. The prognosis is poor in patients with CNS involvement, however using interferon alfa (IFN-a) as treatment the 5-year survival increases to approximately 68% [
3]. Our patient did not have any recent imaging of the brain, so this was not evaluated.
Pathology demonstrates marrow space fibrosis with giant cells, and positive stains for CD68, which is a histiocyte marker. This is in contrary to Langerhans cells, where protein S-100 would be positive. Initially, bone biopsy pathology revealed a non-diagnostic result of marrow space fibrosis. It was only after the bone scan that pathologists were asked to review the samples for ECD, in which case CD68 was found to be positive, confirming the suspicion of ECD. Prognosis is highly variable and depends on the extra-skeletal lesions involved. Due to the variable presentation of ECD, delayed diagnosis is typical, which can lead to high morbidity and mortality rates [
6]. Unfortunately, treatment is not typically successful but includes medications that help control over-production of histiocytes. IFN-a is used as first-line treatment approaches. Currently, BRAF inhibitors including cladribine, anakinra, and vemurafenib are offering promising results and are considered the preferred treatment in more severe cases and in cases that don’t respond to IFN-a [
3]. Vemurafenib has prolonged efficacy in patients with BRAF V600 mutation and is being considered for the new standard of care [
7]. Our patient is currently being managed conservatively with intra-articular injections for symptomatic relief.
Conclusion
ECD is a rare, multisystem disorder that requires knowledge of the disease distribution along with a high degree of clinical suspicion for diagnosis. Biopsy demonstrating histiocytic infiltration and clinical along with radiographic features is imperative for diagnosis. Given the rarity of the disease, referral to an experienced academic center is recommended when diagnosing and treating ECD.
Contributors: CK: manuscript writing, literature review and imaging; RG: manuscript editing, references and imaging. CK will act as a study guarantor. Both authors approved the final version of this manuscript and are responsible for all aspects of the study.
Funding: None; Competing interests: None stated.
References
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681.
- Estrada-Veras JI, O'Brien KJ, Boyd LC, Dave RH, Durham B, Xi L, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1:357-366.
- Diamond EL, Dagna L, Hyman D, Cavalli G, Janku F, Estrada-Veras J. Consensus guidelines for the diagnosis and clinical management of Erherim-Chester disease. Blood. 2014;124:483-492.
- Adawi M, Bisharat B, Bowirrat A. Erdheim-Chester disease (ECD): Case report, clinical and basic investigations, and review of literature. Medicine. 2016;95:e5167.
- Abdellateef EE, Abdelhai AR, Gawish HH, Abdulmonaem GA, Abdelbary EH, Ahmed AI. The first reported case of Erdheim-Chester disease in Egypt with bilateral exophthalmos, loss of vision, and multi-organ involvement in a young woman. Am J Case Rep. 2016; 17:360-370.
- Binyousef RF, Al-Gahmi AM, Khan ZR, Rawah E. A rare case of Erdheim-Chester disease in the breast. Ann Saudi Med. 2017;37:79-83.
- Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, et al. Vemurafenib for BRAF V600–Mutant Erdheim-Chester disease and Langerhans cell histiocytosis. JAMA Oncol. 2018;4:384-388.