6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffa487370000008a06000001000800
6go6ckt5b5idvals|3116
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Sternal cleft (SC) is a rare disorder and comprises 0.15% of all chest wall malformations [
1]. Depending on the degree of separation, SC can be complete or partial, which is further subdivided into superior or inferior SC. Absence of sternum is associated with morbidities including injury to mediastinal viscera, hypothermia, insensible fluid loss and recurrent respiratory infection with its sequelae. It can be an isolated malformation or be associated with a variety of other anomalies. It is imperative to rule out any associated malformations before surgical repair of the defect. There are different methods of SC repair and early intervention during the neonatal period is preferred due to the increased compliance of the thoracic cage leading to successful primary closure.
Case Report
A 25-year-old second gravida with 38 weeks + 4 days gestation presented in labor to our institution and delivered a 2.7 kg female baby vaginally. She had registered herself at our hospital at 15 weeks of gestation for the first time, when she was advised routine antenatal investigations and a level II ultrasound at 18 weeks. Her antenatal blood investigations including diabetes screen was normal. She got a level II ultrasound done at 20 weeks + 3 days which did not show any gross congenital anomaly. The patient was taking antenatal care from the nearby primary health centre as a low risk patient and attended our hospital once in each trimester.
Detailed antenatal history of the mother suggested that she was immunized against tetanus toxoid, and took iron, folic acid and calcium supplements during her pregnancy. There was no history suggestive of any intrauterine infection, radiation exposure, or any drug intake. The mother did not suffer from any chronic illnesses. It was a non-consanguineous marriage and this was her second pregnancy. She had a previous 4-year-old male child delivered vaginally at term. The child had a history of neonatal intensive care unit (NICU) admission due to meconium aspiration and neonatal seizures as a consequence of that. The child was healthy now with normal milestones.
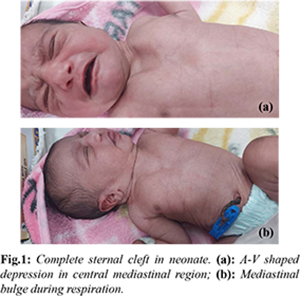
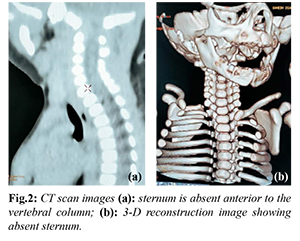
The newborn cried immediately at birth and had an APGAR score of 8, 9, 9 at 1, 5 and 10 minutes. On examination, the newborn had a V-shaped defect of approximately 6×4 cm in the midline. The skin over the defect was normal and the heart pulsations could be palpated under it. The neonate had paradoxical movements of the chest with a bulge in this area during expiration and recession during inspiration [Fig.1a,b]. The neonate was admitted in NICU for observation and further evaluation for probable diagnosis of sternal cleft. The newborn was examined for external hemangiomas, precordial skin tags, cleft lip/palate or supraumbilical median raphe. No such external abnormalities were detected. The newborn underwent X-ray and a computed tomography (CT) imaging of the chest for diagnosis. The chest X-ray suggested an absent sternum with widely spaced medial ends of the clavicles which was confirmed on the CT scan [Fig.2a,b]. Further evaluation for associated cardiac anomalies was done and the echocardiography suggested a congenital acyanotic heart disease with a 2 mm patent foramen ovale (PFO), left to right shunt and normal left ventricular systolic function. The baby also underwent a whole abdomen ultrasound and neurosonogram which were normal. The baby was discharged on 4th postnatal day with advice to follow-up in tertiary centre for surgical correction of the defect which was not available at our institute.
Discussion
Sternal cleft is a rare chest wall anomaly with a reported incidence of 1 in 100,000 live births [
2]. Complete sternal cleft results from failure of midline fusion of the two mesenchymal bars during embryonic development. These two bars form the sternum and fuse between 7th to 10th weeks of gestation. The fusion starts at the manubrium and ends at the xiphoid process caudally; the malformation is therefore classified as superior, inferior or complete depending on the when the fusion process was interrupted [
3]. A superior partial cleft is the most common type accounting for 67% of all patients, followed by the complete form seen in approximately 20%, partial inferior in 11% and sternal foramen in 2.5% [
4]. This neonate had complete absence of sternum which was confirmed radiologically. The etiology of SC is unknown and maternal deficiency of riboflavin or methylcobalamin as well as involvement of HOXb gene have been described as causative factors for SC [
5].
Prenatal diagnosis of the condition is difficult and only around 6% cases of SC are diagnosed prenatally; however, in cases with associated cardiac anomalies, SC can be detected more frequently during the second trimester scan. Majority of patients are diagnosed in the neonatal period when a paradoxical thoracic bulging is noticed in the midline. This is due to the protrusion of the mediastinal viscera during expiration. In this case also, the second trimester ultrasound did not show any anomaly and failed to pick up the absence of sternum. SC in our case was noted at birth due to the mediastinal bulge and paradoxical movements of the chest of the neonate. SC may be associated with other anomalies like maxillo-facial hemangiomas, cleft lip/palate, gastroschisis, precordial skin tags and supraumbilical raphe which can be ruled out by a careful physical examination of the neonate. SC has also been found to be associated with pentalogy of Cantrell (ectopia cardis, sternal cleft, intracardiac defects, omphalocele and pericardial defect), VACTERL syndrome and PHACES syndrome (posterior fossa abnormalities, hemangioma, arterial abnormalities, coarctation of aorta, eye abnormalities and sternal malformations) [
6,
7]. Imaging studies like chest X-ray, CT scan, ECG and echocardiography are important diagnostic tools to identify associated anomalies. Our patient had the diagnosis confirmed on CT scan of the chest; abdominal ultrasound and neurosonogram were normal while the echocardiogram detected a small PDA.
Early detection and surgery of SC is critical as paradoxical breathing in such neonates leads to impaired gas exchange and inability to clear secretions which further causes frequent chest infections and its sequelae [
8]. Absence of sternum can also cause hypothermia, insensible fluid loss and there are is always a danger of life-threatening injury to the mediastinal viscera and vessels [
4]. Surgical correction should be performed during the neonatal period when the thoracic wall is highly complaint and primary closure can be achieved without any cardiopulmonary compromise [
9]. Different methods of surgical repair include primary approximation, sliding or rotating chondrotomies and reconstruction with autogenous or prosthetic tissues [
10]. In female patients it is important to be careful not to damage the mammary glands so that normal breast development occurs later. With increasing age, not only does the chest wall get less elastic but also the venous return and lung compliance is decreased leading to complications and poor surgical outcomes [
8]. Even after surgical correction, patients can develop other chest wall deformities like pectus excavatum and therefore a strict follow-up is recommended.
Conclusion
SC is a rare congenital malformation which can either be isolated or occur in association with other anomalies and genetic syndromes. It is easily diagnosed in the neonatal period due to the obvious paradoxical movements of the thorax. Absence of sternum is associated with significant morbidities including hypothermia, recurrent respiratory infections and injury to mediastinal viscera and vessels. Surgical repair is the treatment of choice which should ideally be performed in the early neonatal period.
Contributors: SM, AG: manuscript writing, patient management; PS, NM: manuscript editing, patient management; JP: critical inputs into the manuscript and imaging. SM will act as a study guarantor. All authors approved the final version of this manuscript and are responsible for all aspects of this study.
Funding: None; Competing interests: None stated.
References
- Acastello E, Majluf R, Garrido P, Barbosa LM, Peredo A. Sternal cleft: a surgical opportunity. J Pediatr Surg. 2003;38(2):178-183.
- Ngoc Thach P, Tran Ban H, Anh Dung HV, Minh Chieu V, Viet Tanh NT, Truong Nhan V, et al. A case report of surgical repair of cleft sternum in a child. Clin Ter. 2021; 172(6):495-499.
- Zamfir C, Zamfirescu A, Tanase C, Basca I. Sternal cleft- A rare congenital malformation. Ped Surg Case reports. 2014;2:97-100.
- Torre M, Rapuzzi G, Carlucci M, Pio L, Jasonni V. Phenotypic spectrum and management of sterna cleft: literature review and presentation of a new series. European J Cardio-Thoracic Surgery. 2012;41:4-9.
- Engum SA. Embryology, sternal clefts, ectopia cordis, and Cantrell’s pentalogy. Semin Pediatr Surg. 2008;17(3):154-160.
- Shrivastava A, Siddiqui SA. Congenital absence of sternum in an infant. BMJ Case Rep. 2017;7:bcr2016219081.
- Dehdashtian M, Peyvasteh M, Emami-Moghaddam A. VACTERL association with sternal cleft. Pak J Med Sci. 2007;23:790e1.
- Mazzie JP, Lepore J, Price AP, Driscoll W, Bohrer S, Perlmutter S, et al. Superior sterna cleft associated with PHACES syndrome: Postnatal sonographic findings. J Ultrasound Med. 2003;22:315-319.
- Trivedi PM, Jagannathan R, Jagannathan N. Congenital absence of the sternum in a neonate. Anesthesiology. 2014;120:752.
- Semlacher RA, Nuri MAK. Successful management of absent sternum in an infant using porcine acellular dermal matrix. Arch Plast Surg. 2019;46(5):470-474.