6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff68df38000000a706000001000300
Introduction
Apert syndrome is a genetic disorder with autosomal dominant inheritance and is caused by FGF (fibroblast growth factor) receptor-2 gene mutations [1]. The entity has been named after a French doctor Eugene Apert who first described a case series of nine patients with Apert syndrome in 1906 [2]. This rare syndrome is characterised by craniofacial deformities and malformations of hands and feet and is also known as acrocephalosyndactyly. ‘Acrocephalo’ stands for tall and peaked skull and is caused by bi-coronal synostosis (premature fusion of bilateral coronal sutures). ‘Syndactyly’ stands for fused digits of hands and feet. Craniofacial deformities specifically seen in this syndrome include midface hypoplasia, hypertelorism, proptosis and a beaked nose [1]. CNS malformations are associated and include abnormalities of corpus callosum including hypoplasia and aplasia, cavum vergae, hypoplasia of septum pellucidum and posterior fossa arachnoid cysts [3].
Case Report
A one-year-old female child was brought by her parents to the Paediatrics outpatient department with complaints of delayed milestones, abnormal facies and fused fingers and toes of hands and feet since birth. A complete medical history was obtained. The antenatal period was uneventful with no history of any teratogenic or radiation exposure during the antenatal period. Birth history was normal with normal cry at birth. No parental consanguinity or similar complaints were seen in the siblings. Clinical examination revealed dysmorphic facial features in the form of flat facies, ocular hypertelorism, beaked nose, prominent forehead and elongated face and skull (acrocephaly) and fused digits of hands and feet [Fig.1]. No cleft/ pseudo-cleft lip or palate was noted in the patient. Non-contrast computed tomography of head and radiograph of extremities and spine were taken for further evaluation. Radiographs of the feet revealed bilateral soft tissue syndactyly, single dysplastic and broadened phalanx of bilateral great toes and presence of only two phalanges in 2nd-5th toes. Syndactyly (bony and soft tissue) were noted in both hands [Fig.2]. Radiograph of the spine found no obvious abnormality with no evidence of segmentation anomaly or dysplastic features. Radiographs of thighs and legs were unremarkable [Fig.3].
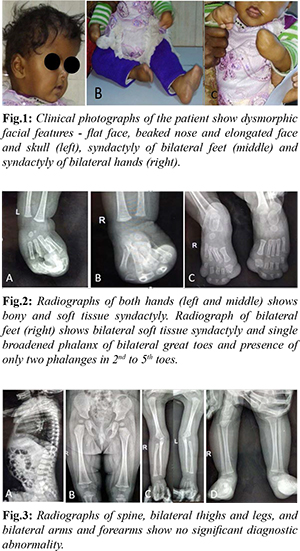
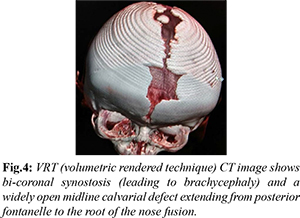
VRT (volumetric rendered technique) image generated from non-contrast CT of the head revealed prominent convolutional markings, bicoronal synostosis (leading to brachycephaly) and a widely open midline calvarial defect extending from posterior fontanelle to the root of the nose [Fig.4]. A note was made of hypoplasia of bilateral maxillae. Brain parenchymal abnormalities were also present with evidence of thinning of corpus callosum, absent cavum septum pellucidum, ventriculomegaly and enlarged posterior fossa cystic space (likely mega cisterna magna) [Fig.5].
Based on the clinic-radiological abnormalities, acrocephalosyndactyly syndrome was considered as a broad differential diagnosis. Various syndromes with overlapping features come under this entity and include Apert syndrome, Apert-Crouzon syndrome, Saethre-Chotzen syndrome, Goodman syndrome, Pfeiffer syndrome and Robinow-Sorauf syndrome. Syndactyly essentially differentiates Apert from Crouzon’s syndrome. Enlarged thumbs and great toes are typically seen in Pfeiffer syndrome and helps differentiate it from Apert syndrome [4]. Skeletal dysplasias were essentially ruled out after examination of radiographs of spine and extremities. Presence of classical triad of craniosynostosis, hypoplasia of mid face and symmetric syndactyly of the feet and hands favoured the radiological diagnosis of Apert syndrome. Genetic testing was advised to the patient but the patient differed because of money constraints.
The patient was referred to Plastic Surgery and underwent surgery for syndactyly. Soft tissue syndactyly was released in both hands, and was then referred to higher centre for further staged reconstruction procedures.
Discussion
Acro-cephalo-syndactyly type 1 or Apert syndrome is a rare genetic syndrome with autosomal dominant inheritance and presents with a classical triad of craniosynostosis, hypoplasia of mid face and symmetrical syndactyly of the feet and hands [1]. Commonly associated central nervous system malformations include corpus callosal dysgenesis, septum pellucidum abnormalities and hydrocephalus [3]. The entity was reported for the first time in the year 1894 by S. W. Wheaton [5]. Dr Eugene Apert, a French paediatrician described a case series of nine patients with Apert syndrome in 1906 [2]. Mutations of FGFR2 (fibroblast growth factor receptors) on chromosome 10q are implicated. Mutations in sutural progenitor cells render them unable to transduce signals from FGFs (fibroblast growth factors) and produce the fibrous component necessary for development of normal suture [6].
The estimated rate of incidence of Apert syndrome is 1:160000. No sex predilection is seen with male and female children being affected equally. Increasing paternal age is associated with higher incidence rates. [7]. The clinical features described are craniofacial dysmorphism, protrusion of eyeballs, high arched palate, bifid uvula and syndactyly [8]. Developmental delay is often present. The most common calvarial abnormality is bilateral coronal synostosis leading to shortened anterior-posterior diameter of skull. Wide open midline calvaria defect extending from posterior fontanelle to the root of the nose is noted in place of normal anterior fontanelle and sagittal-metopic sutures [9]. There is defect in the normal formation of sutures and instead, bone islands appear within the defect, which eventually coalesce by about 36 months. Intracranial anomalies are seen in almost half of the patients with Apert syndrome. Corpus callosal dysgenesis, hydrocephalus, and agenesis of septum pellucidum are reported in 25-30% of the patients. Cavum vergae and arachnoid cysts are reported in 10-12% of the cases. Other rare intracranial manifestations are Arnold Chiari I malformation and venous anomalies [10,11].
Sphenoid-ethmoidal and the frontal-ethmoidal sutures fuse early, leading to short cranial base resulting in hypoplastic midface. Increased interocular distance as well as hypoplasia of bilateral maxillae result in mid face retrusion with protrusion of eye balls. Bilaterally symmetric hand and feet soft tissue and bony syndactyly is characteristic [12,13]. The hands and feet deformities help to distinguish it from other craniosynostoses syndromes. Severe syndactyly and fusion of phalanges may give appearance of mitten’ or ‘hoof’ hands. Short radially deviated thumb and dislocation of radial heads may be present [13]. There is progressive fusion of phalanges, carpals and tarsals and large joints. Cervical spine anomalies may be seen in some cases with progressive fusion of cervical spine (mostly at the level of C5/C6) [11].
Computed tomography is a sensitive imaging modality in detecting any underlying structural abnormality in calvaria and brain. CT is also helpful to assess the nasopharyngeal airway as craniofacial abnormalities may contribute to airway obstruction. Associated choanal atresia and nasal septal deviation can be detected on CT. Recognition of characteristic clinical and imaging abnormalities are crucial to radiologists and clinicians in the diagnosis and management of these patients as well as to assess the prognosis. Since there is multi-system involvement, a multidisciplinary approach with combined efforts from neurosurgery, oral and maxillofacial surgery, ophthalmic and plastic surgery team is needed in the management of patients with Apert syndrome [14]. Timing of surgical intervention may improve the overall prognosis and patient’s cognitive development. Surgeries for correction of cranio-facial abnormalities as well as separation of connected fingers and toes form a part of the treatment [13]. VP shunting is less frequently required [10].
Conclusion
Apert syndrome is a rare acro-cranio-synostosis syndrome characterised by syndactyly and craniosynostosis. Imaging is helpful in recognition of calvarial and facial bony abnormalities as well as associated central nervous system abnormalities and is crucial in the management of these patients. Surgeries for correction of cranio-facial abnormalities as well as separation of connected fingers and toes form part of the treatment.
Contributors: SS, MM drafted the work and revised it critically with the help of SW and MA. MM will act as a study guarantor. All authors approved the final version of this manuscript and are responsible for all aspects of this study.
Funding: None; Competing interests: None stated.
References
- Ileri Z, Goyenc YB. Apert syndrome: A case report. Eur J Dent. 2012;6:110-113.
- Apert E. DeL’acrocéphalosyndactylie. Bulletins et mémoires de la Sociétémedicale des hôpitaux de Paris.1906;23:1310.
- Bhatia PV, Patel PS, Jani YV, Soni NC. Apert's syndrome: Report of a rare case. J Oral Maxillofac Pathol. 2013;17:294-297.
- Martelli H, Paranaiba LM, Miranda RT, Orsi J, Coletta RD. Apert syndrome: Report of a case with emphasis on craniofacial and genetic features. Pediatr Dent. 2008;30:464-468.
- Wheaton SW. Two specimens of congenital cranial deformity in infants associated with fusion of the fingers and toes. Transactions of the Pathological Society of London. 1894;45:238.
- Rynearson RD. Case Report: Ortho dontic and dento facial orthopedic considerations in Apert’s syndrome. Angle Orthod. 2000;70:247-252.
- Goriely A, McVean GA, Röjmyr M, Ingemarsson B, Wilkie AO. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643646.
- Canpolat M, Büyükayhan D, Günes T, Akçakus M, Öztürk A, Kurtoglu S. Apert Syndrome: A Case Report. Erciyes Med J. 2009;31:53-61.
- Kreiborg S, Cohen MM. The infant Apert skull. Neurosurg Clin N Am. 1991;2:551-554.
- Munarriz PM, Pascual B, Castaño-Leon AM, García-Recuero I, Redondo M, Aragón AM, et al. Apert syndrome: Cranial procedures and brain malformations in a series of patients. Surg Neurol Int. 2020;29:361.
- Breik O, Mahindu A, Moore MH, Molloy CJ, Santoreneos S, David DJ. Central nervous system and cervical spine abnormalities in Apert syndrome. Childs Nerv Syst. 2016;32:833-838.
- Koca TT. Apert syndrome: A case report and review of the literature. North Clin Istanb. 2016;3:135-139.
- Pettitt DA, Zeeshaan A, Mishra A, McArthur P. Apert syndrome: A consensus on the management of Apert hands. Journal of Cranio-Maxillofacial Surgery. 2017;45:223-231.
- Fadda MT, Lerardo G, Ladniak B, Di Giorgio G, Caporlingua A, Raponi I, et al. Treatment timing and multidisciplinary approach in Apert syndrome. Ann Stomatol (Roma). 2015;6:58-63.