|
|
|
|
|
Pleuroparenchymal Fibroelastosis following Asbestos Exposure
|
|
|
|
Neha Bipin Patel1, Timothy Patrick McCann1, David William Wiltse2 Departments of 1Internal Medicine and 2Pulmonary Medicine, Good Samaritan Hospital, Cincinnati, OH, United States. |
|
|
|
|
|
Corresponding Author:
|
|
Dr Neha Bipin Patel Email: neha_patel@trihealth.com |
|
|
|
|
|
|
|
|
Received:
16-FEB-2022 |
Accepted:
16-OCT-2022 |
Published Online:
05-NOV-2022 |
|
|
|
|
|
|
|
Abstract
|
|
|
|
Background: Pleuroparenchymal fibroelastosis is considered a rare form of interstitial lung disease. There are a growing number of case reports, however no large-scale studies detailing both clinical and histopathological features along with long term follow-up. An association with asbestos exposure has been described however this too has been limited to a few case reports. Case Report: A 73-year-old man with a history of interstitial lung disease and asbestos exposure presented with dyspnea with biopsy and imaging suggestive of pleuroparenchymal fibroelastosis. Despite steroid and anti-fibrotic therapy, the disease advanced rapidly causing the patient and family to pursue palliative care. Conclusion: Asbestos exposure is a significant risk factor for pleuroparenchymal fibroelastosis, a rare but increasingly recognized entity. Biopsy and image findings are the mainstay of diagnosis, pulmonary function tests are useful in prognostication. Treatment is supportive. |
|
|
|
|
|
Keywords :
|
Acute Respiratory Distress Syndrome, Asbestosis, Interstitial Lung Disease, Pulmonary Fibrosis, Diffuse Parenchymal Lung Disease.
|
|
|
|
|
|
|
|
|
|
|
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff24c038000000a406000001000c00 6go6ckt5b5idvals|3132 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
The American Thoracic Society categorizes pleuroparenchymal fibroelastosis (PPFE) as a diffuse parenchymal lung disease (DPLD) and is considered a rare form of interstitial lung disease (ILD) [ 1]. Distinguishing characteristics include upper-lobe predominant fibrosis and pleural thickening along with unique histopathologic features of collagenous fibrotic deposition within intra-alveolar space and visceral pleural as well as subpleural elastosis [ 1]. These two distinctive characteristics, a predilection for upper lobe disease and fibrosis related to elastic fiber proliferation, separate it from the usual collagenous fibrosis seen after lung parenchymal injury. PPFE has been compared to Amanti’s disease, a similar disease process of idiopathic pulmonary upper-lobe-localized fibrosis described by Japanese investigators. While the clinical features of those with Amanti’s disease appear to be similar to those described with PPFE, there is still a debate whether the two are the same disease process [ 2]. PPFE was first described in 2004 with over 110 cases have been reported in the literature to date [ 2]. The true incidence and prevalence of PPFE are not known, owing to uncertainties in its detection, misdiagnosis, and the absence of agreed criteria for its identification [ 3]. There are a growing number of case reports, however, there are no large-scale studies detailing both clinical and histopathological features along with long term follow-up. An association with asbestos exposure has been described however this too has been limited to a few case reports [ 4- 6]. Here we present a case of PPFE in an elderly male with a childhood history of significant asbestos exposure.
Case Report
A 73-year-old man with a history of coronary artery bypass graft, chronic interstitial lung disease, and childhood asbestos exposure presented to the emergency department with acute onset cough, diminished cough reflex, and worsening chronic dyspnea. Positive review of symptoms included fatigue and generalized weakness. He has reported chronic dyspnea since age 62. Vitals on presentation were significant for 37.8 Celsius temperature, tachycardia 108 beats per minute, tachypnea 23 respirations per minute, and pulse oximetry of 92% SpO2 on room air. Oxygen saturation initially improved to 99% on 2 liters via nasal cannula. His body mass index was 19.79 kg/m2. Respiratory exam exhibited rapid shallow breaths and fine bibasilar crackles on lung auscultation. The remainder of the exam was unremarkable. Initial laboratory results showed complete leukocyte count 7.4 thousand/mcL (3.6-10.5 thousand/mcL), lactic acid 2.0 mmol/L (0.5-2.0 mmol/L), and procalcitonin 6.26 ng/mL (<0.10 ng/mL). Radiographic images of the chest (CXR) prompted further evaluation with computed tomography (CT) [Fig.1,2]. Given the findings concerning bacterial pneumonia, the patient was started on empiric intravenous (IV) antibiotic therapy for bacterial pneumonia with ceftriaxone 2g and azithromycin 500 mg daily. Further pertinent infectious work up included a negative methicillin-resistant staphylococcus aureus (MRSA) nasal polymerase chain reaction (PCR), a negative covid-19 viral nasal PCR, and a positive respiratory syncytial virus (RSV) nasal PCR.
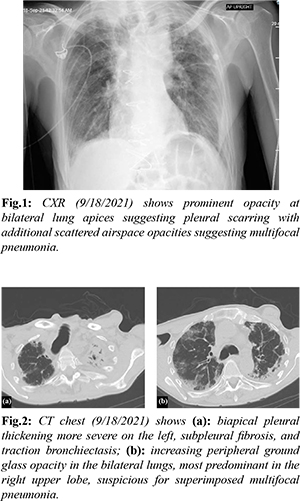
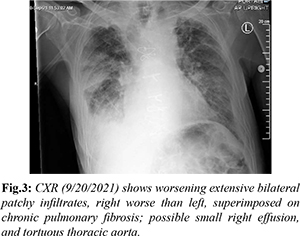
Over the next 24 hours, he developed hypotension, tachycardia, and severe acute respiratory distress syndrome requiring 55% fraction of inspired oxygen (FiO2) at 50 L/min via high flow nasal cannula. Repeat CXR showed worsening extensive bilateral patchy infiltrates [Fig.3]. Treatment was escalated to broad spectrum IV antibiotics piperacillin-tazobactam 4.5 g every 8 hours, antiviral ribavirin 600 mg every 12 hours, and prednisone 20 mg daily along with admission to the intensive care unit. Due to minimal improvement despite aggressive treatment, the decision was made to pursue comfort care measures and he expired on day nine of admission.
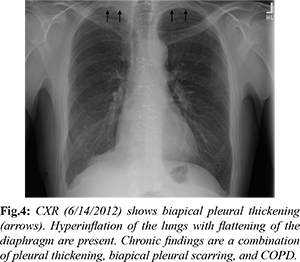
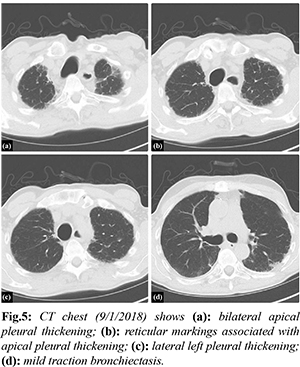
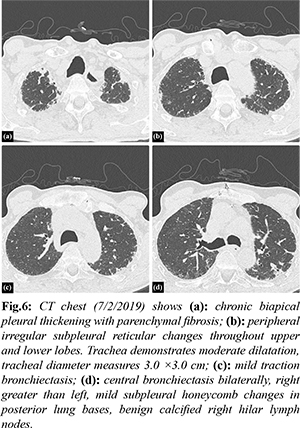
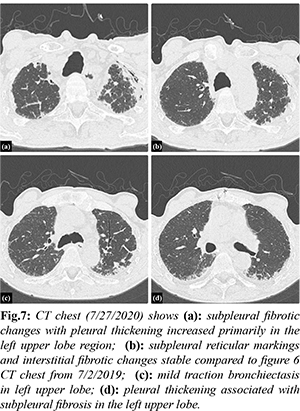
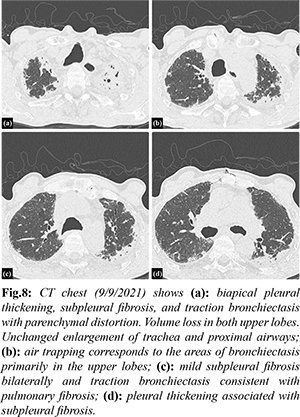
Investigation into his chronic pulmonary disease revealed an unusual process first noted in 2011. Initially, he developed recurrent exudative bilateral pleural effusions one year after coronary artery bypass graft. Extensive malignant, infectious, rheumatologic, and autoimmune work up was unremarkable. Right pleural biopsy revealed chronic pleuritis, fibrous pleural tissue, and lymphoplasmacytic infiltrate of the parenchyma. Beginning in 2012 there was evidence of chronic biapical pleural thickening on CXR [Fig.4]. Periodic imaging since then depicted progression of pleural thickening, subpleural fibrosis, and reticular changes more prominent in bilateral upper lobes with evidence of traction bronchiectasis as seen on chest CT from 2018, 2019, 2020, 2021 [Fig.5-8].
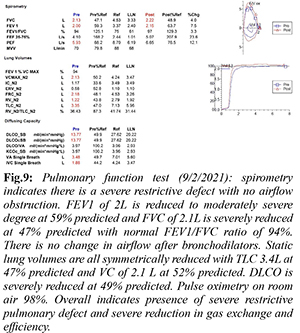
A 2019 pulmonary function test (PFT) reported a moderate restrictive lung defect in a pattern consistent with parenchymal restriction. Spirometry showed forced expiratory volume in 1 second (FEV1) 2.47L at 68% predicted, forced vital capacity (FVC) 2.53L at 51% predicted with a FEV1/FVC ratio of 98%. All static lung volumes were moderately reduced. Diffusion capacity of carbon monoxide (DLCO) was mildly reduced at 73% predicted. When compared to a 2021 PFT performed prior to final admission, there was evidence of disease progression with a severe restrictive defect and severe reduction in gas exchange and efficiency [Fig.9]. Spirometry showed FEV1 of 2L reduced to 59% predicted and FVC of 2.1L severely reduced to 47% predicted with a FEV1/FVC ratio of 94%. DLCO was severely reduced at 49% predicted.
Given primarily upper lobe disease progression on imaging in addition to progressive restrictive pulmonary defects with decreased diffusion capacity, the condition appeared atypical for usual interstitial pneumonia and more consistent with PPFE. Despite therapy with prednisone 4 mg daily and pirfenidone 267 mg twice daily, the disease advanced rapidly causing weight loss, refractory dyspnea, and increased susceptibility to infections which ultimately led him to the hospital admission described.
Discussion
Patients presenting with PPFE exhibit chronic progressive dyspnea with associated symptoms of cough and non-specific symptoms of generalized weakness and fatigue [ 3]. Physical exam findings include tall, slender stature and flattened thoracic cage which refers to a low anteroposterior chest diameter to transverse chest diameter ratio likely a result of progressive upper lung volume loss [ 7]. Bilateral coarse crackles are heard on lung auscultation [ 7]. All presentations and findings are similar to our patient. PPFE is diagnosed based on radiological and histological findings [ 1]. Characteristic radiographic findings include upper lobe predominant fibrosis, pleural fibrosis, as well as traction bronchiectasis. Flattened thoracic cage and elevated hilar opacities due to traction from upper lobe fibrotic opacities can be seen on CXR [2]. CT chest findings in the early stages of the disease can present as subpleural nodular and reticular opacities in the apex with sparing of the middle and lower lobes. Advance stage findings present as fibrotic upper lobe opacities with traction bronchiectasis extending to adjacent lobes [ 2]. Our patient’s annual chest CT imaging [Fig.5-8] demonstrates these characteristic findings and the noted progression of disease. Initial findings on histopathology are defined by pleural and subpleural fibrosis with fibroelastosis predominating over collagenous fibrosis [ 7]. It affects lung tissue closer to the pleura. The parenchyma cytopathology shows lymphoplasmacytic infiltrate and fibroblastic foci [ 5]. Performing elastic fiber stains may aid in deciphering fibroelastosis versus collagenous fibrosis by showing elastic band-like pleural thickening [ 4]. As the disease progresses, the lower lobes may become more involved however the pathology may involve non-PPFE patterns [7]. Medical discussion should include consideration for biopsy if feasible early in the disease course [ 7]. Our patient’s pleural biopsy revealed chronic pleuritis, fibrous pleural tissue, and lymphoplasmacytic infiltrate of the parenchyma, all similar findings noted in PPFE. With or without biopsy, pulmonary function tests [PFTs] can aid in the diagnosis [ 8]. PFTs will usually show restrictive defects with low TLC and FVC, normal FEV1/FVC, yet preserved RV and increased RV/TLC ratio [ 8]. Diffusion capacity is generally normal in early stages however progressively decreases as the disease advances [ 8]. Our patient’s PFT results from 2019 and again in 2021 [Fig.9] demonstrates these results. The progression seen from the PFT from 2019 to 2021 is consistent with ILD and pulmonary fibrosis. There is no airflow obstruction present. Ventilatory reserve and oxygenation are remarkably normal despite the severe restrictive defect. The DLCO was decreased, likely given the severity of disease. Treatment options are minimal including steroids, anti-fibrotic medications, and immunosuppressive agents based on prior documented case reports, yet none have proven to significantly change the disease course [ 7]. Our patient was treated with prednisone and pirfenidone with no significant blunting of disease course. There have been reports of patients with PPFE receiving lung transplantation however follow up after the transplantation has been limited [ 9]. PPFE is categorized as either idiopathic or secondary. Secondary PPFE arises from underlying disease processes such as lung transplantation, hematopoietic stem cell transplantation [HSCT], genetics, chemotherapy and radiation therapy, or occupational exposure [ 2]. In a retrospective review of high-resolution computed tomography (HRCT) of 53 lung transplantation patients and 700 HSCT patients, radiographic evidence of PPFE was found in 4 (7.5%) lung transplantation patients and 2 (0.28%) HSCT patients [ 10]. Another study proposed autoimmune disease as a possible contributor in the pathogenesis of PPFE after five of twelve patients with PPFE were positive for rheumatoid factor (RF), antinuclear antibody, and anti-double-stranded deoxyribonucleic acid (anti-dsDNA). Genetics may also play a role. Family history of pulmonary fibrosis has been associated with diagnoses of PPFE [ 11]. A case series of three sisters with PPFE revealed significant family history of ILD [ 12]. Mutations in genes of telomere maintenance, including telomerase reverse transcriptase (TERT) and telomerase ribonucleic acid (RNA) component (TERC) were highly associated (10%) with ILD caused by PPFE [ 13, 14]. Chemotherapy has also been implicated as a cause. A case series of six patients in France showed significant history of exposure to alkylating agents, specifically cyclophosphamide [ 5] and carmustine [ 1, 15]. Occupational exposures related to inhaled toxicants including dust, asbestos, and aluminium have been reported to be linked to developing PPFE [ 16]. Asbestos specifically is known to induce a fibro-elastotic interstitial tissue response rather than fibrotic tissue response along with pleural thickening suggesting asbestos exposure as a plausible direct cause of PPFE [ 2]. The association with asbestos has been limited to case reports. PPFE was found in a 59-year-old female with childhood asbestos exposure, however she had also had exposure to alkylating agents [ 4]. A 28-year-old plumber who had been chronically exposed to asbestos presented with worsening dyspnea and radiological and biopsy evidence of PPFE [ 5]. Another case was reported in a patient after long term exposure to inhaled occupational agents including asbestos and silica [ 6]. Given that only minimal case reports document the link between asbestos exposure and PPFE, we aim to add to this literature. Our patient’s history of asbestos exposure as a child is likely a major contributing agent for the fibroelastic proliferative tissue response and is likely a rare, possibly under-reported sequela of asbestos exposure. For a decade, the etiology of our patient’s chronic interstitial lung disease remained unclear leading to progressively worsening symptoms and ultimately leading to increased susceptibility to community acquired infections and his final hospitalization. When biopsy was performed in 2010, the concept of PPFE was still in its infancy, only first described in 2004. Unfortunately, we were unable to obtain pathology images given the age of the specimens, so we are left with only the pathologist’s description. As such, we are unable to perform additional stains such as elastic fiber stains which would strengthen the diagnosis of PPFE and the correlation between the patient’s childhood asbestos exposure. The periodic imaging and PFTs help strengthen the diagnosis, despite the lack of further pathology investigations.
Conclusion
PPFE is a rare but increasingly recognized entity. Asbestos exposure is associated with development of PPFE. Imaging and biopsy findings are the mainstay for diagnosis. Current treatment is supportive with infection risk mitigation and symptomatic management. Large scale studies are needed to further characterize risk factors and possible treatments.
Contributors: NBP: Conception of the work and design of the work, acquisition of data, analysis of data and interpretation of data. Also significantly contributed to drafting and revising the work critically for important intellectual content; TPM, DWW: contributed to conception of the work and design of the work and contributed to revising the work critically for important intellectual content. NBP will act as a study guarantor. All authors approved the final version of this manuscript and are responsible for all aspects of this study. Funding: None; Competing interests: None stated.
References - Travis, WD, Costabel, U, Hansell, DM, King, TE, Lynch, DA, Nicholson, AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Resp Crit. 2013;188(6):733-748.
- Watanabe K. Pleuroparenchymal fibroelastosis: Its clinical characteristics. Curr Respir. 2013;9(4):229-237.
- Chua, F, Desai, SR, Nicholson, AG, Devaraj, A, Renzoni, E, Rice, A, et al. Pleuroparenchymal fibroelastosis. A review of clinical, radiological, and pathological characteristics. Ann Am Thorac Soc. 2019;16(11):1351-1359.
- Becker, C, Gil, J, Padilla, M. Idiopathic pleuroparenchymal fibroelastosis: an unrecognized or misdiagnosed entity? Mod Pathol. 2008;21:784-787.
- Piciucchi, S, Tomassetti, S, Casoni, G, Sverzellati, N, Carloni, A, Dubini, A, et al. High-resolution CT and histological findings in idiopathic pleuroparenchymal fibroelastosis: features and differential diagnosis. Respir Res. 2011;12(1):111.
- Xu L, Rassaei N, Caruso C. Pleuroparenchymal fibroelastosis with long history of asbestos and silicon exposure. Intern J Surg Path. 2018;26(2):190-193.
- Bonifazi, M, Montero, MA, Renzoni, EA. Idiopathic pleuroparenchymal fibroelastosis. Curr Pulm R. 2017;6(1):9-15.
- Watanabe, K, Nagata, N, Kitasato, Y, Wakamatsu, K, Nabeshima, K, Harada, T, et al. Rapid decrease in forced vital capacity in patients with idiopathic pulmonary upper lobe fibrosis. Respir Invest. 2013;50(3):88-97.
- Ali, MS, Ramalingam, VS, Haasler, G, Presberg, K. Pleuroparenchymal fibroelastosis [PPFE] treated with lung transplantation and review of the literature. BMJ Case Rep. 2019;12(4):e229402.
- Mariani, F, Gatti, B, Rocca, A, Bonifazi, F, Cavazza, A, Fanti, S, et al. Pleuroparenchymal fibroelastosis: the prevalence of secondary forms in hematopoietic stem cell and lung transplantation recipients. Diagn Interv Radiol. 2016;22(5):400-406.
- Reddy TL, Tominaga M, Hansell DM, von der Thusen, J, Rassl, D, Parfrey H, et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012;40(2):377-385.
- Azoulay, B, Paugam, MF, Heymann, M, Kambouchner, A, Haloun, D, Valeyre, JP, et al. Familial extensive idiopathic bilateral pleural fibrosis. Eur Respir J. 1999;14(4):971-973.
- Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48:1710-1720.
- Nunes, H, Jeny, F, Bouvry, D, Picard, C, Bernaudin, JF, Ménard, C, et al. Pleuroparenchymal fibroelastosis associated with telomerase reverse transcriptase mutations. Eur Respir J. 2017;49(5):1602022.
- Beynat-Mouterde, C, Beltramo, G, Lezmi, G, Pernet, D, Camus, C, Fanton, A, et al. Pleuroparenchymal fibroelastosis as a late complication of chemotherapy agents. Eur Respir J. 2014;44(2):523-527.
- Huang Z, Li S, Zhu Y, Zhu H, Yi X. Pleuroparenchymal fibroelastosis associated with aluminosilicate dust: a case report. Int J Clin Exp Pathol. 2015;8(7):8676-8679.
|
|
|
|
|
|
|
Search Google Scholar for
|
|
|
Article Statistics |
|
Patel NB, McCann TP, Wiltse DWPleuroparenchymal Fibroelastosis following Asbestos Exposure.JCR 2022;12:106-112 |
|
Patel NB, McCann TP, Wiltse DWPleuroparenchymal Fibroelastosis following Asbestos Exposure.JCR [serial online] 2022[cited 2026 May 21];12:106-112. Available from: http://www.casereports.in/articles/12/4/Pleuroparenchymal-Fibroelastosis-following-Asbestos-Exposure.html |

|
|
|
|
|