Introduction
In developing countries like India, most common cause of anaemia is nutritional; vitamin B12 and folic acid deficiency leading to megaloblastic anaemia is one of the common patterns. Typically, vitamin B12 deficiency is characterised by pancytopenia and ineffective erythropoiesis leading to intravascular hemolysis. The main feature differentiating megaloblastic and haemolytic anaemia is the erythropoietic response of bone marrow resulting in increased reticulocyte count, a test too often neglected in initial workup of a patient with anaemia. Moreover, pancytopenia is generally not a feature of hemolytic anaemia, except in advanced cases associated with hypersplenism and PNH.
A 35 year old vegan male presented with yellow discoloration of sclera and skin since last 3 months and acute abdominal pain since one week. He had been treated for megaloblastic anaemia with intravenous therapy. During evaluation, he had pancytopenia, raised MCV, reticulocytosis, indirect hyperbilirubinemia, elevated LDH and ultrasound abdomen during the episode of pain abdomen was suggestive of ischemic colitis. CT angiography showed mesenteric vein thrombosis. He was evaluated for paroxysmal nocturnal hemoglobinuria (PNH) as he had classical triad of pancytopenia, hemolysis and venous thrombosis. Eventually, patient was found to be positive for PNH clone.
Case Report
A 35 year old male was admitted with chief complaints of yellowish discoloration of sclera and skin since last 3 months. It was gradual in onset, progressive and associated with high yellow coloured stools. There was no change in colour of urine. It was preceded by single episode of fever, lasting 3 to 4 days. He also had generalised weakness, light headedness, and giddiness since 7 months. The weakness was progressive to the extent that he couldn’t walk without support during the third month of illness. One week back, patient also got severe abdominal pain, which was associated with abdominal distension and nausea but pain subsided two days back. He had no history of syncope, anorexia, palpitations, tinnitus, tingling sensation of extremities, worms in stool or pica. No history of chronic blood loss, malabsorption, exposure to drugs/ chemicals/ radiation, any chronic illness or similar history in past. He is married and has two children. He is a vegetarian, non smoker, non alcoholic, and tobacco chewer since, last 20 years. His mother died of aplastic anaemia 20 years back, while elder sister died of AML 15 years back. He had got admitted to various hospitals for his illness, and was treated for megaloblastic anaemia by intravenous therapy. He was had been transfused with 8 units of blood in the last 3 months.
On examination, his vitals were stable. He was having severe pallor, icterus and lemon yellow skin colour. There was no lymphadenopathy, glossitis, chelitis, frontal bossing, malar prominence, sternal tenderness, pedal edema or leg ulcers. On systemic examination, he had non-tender hepatomegaly (liver span of 18 cm in right mid-clavicular line) and splenomegaly (3 fingers breadth below left costal margin towards umbilicus). Since he had anaemia with jaundice and mild hepatosplenomegaly, provisional diagnosis of haemolytic anaemia was kept. During hospital stay, patient again got severe abdominal pain, which was associated with nausea and brownish discoloration of urine.
On investigating it was seen that the patient had severe anaemia (Hb 5.1 gm%), leucopenia (TLC 3200/cu mm) and thrombocytopenia (90,000/cu mm). Total RBC count was 1.34 million/ cu mm with myelocyte count of 3%. Nucleated RBCs (3/100 WBC) were present. There was reticulocytosis (8%); MCV was raised (108.3 fL). RBCs were macrocytic and normochromic with polychromasia, and reduced platelets.
There was indirect hyperbilirubinemia (4.0 mg%), with total bilirubin of 4.9 mg%. SGOT/SGPT was normal (20/18). Kidney functions and serum electrolytes were within normal limits. Serum LDH was very high (14,803), while iron profile was suggestive of iron deficiency.
Ultrasound abdomen confirmed hepatospleno-megaly (liver 15.3 cm, with normal echotexture; spleen 13.2 cm, normal echogenicity). A repeat ultrasound was done for abdominal pain which showed moderate ascites with thickened distal transverse colon wall (1.9 cm) & possibility of ischemic colitis was kept. Bone marrow aspirate suggested of cellular bone marrow; megaloblastic erythroid hyperplasia with dyserythropoesis. Myeloid cells were having adequate maturation; megakaryocytes were adequate in number.
Investigations were in favour of advanced megaloblastic anaemia, with ineffective erythropoiesis and intravascular hemolysis (iron deficiency and serum LDH in thousands). But patient had already received intravenous methylcobalamin and folic acid therapy. On investigation, we found his serum vitamin B12 and folic acid levels to be normal (serum vitamin B12 = 450 pg/mL and serum folic acid 19 ng/mL). Intravascular hemolysis was confirmed by plasma haemoglobin levels (raised, 8.5 mg%) and urinary hemosiderin levels (three times positive). Moreover, serum haptoglobin levels were also low.
A repeat ultrasound was done for abdominal pain which showed moderate ascites with thickened distal transverse colon wall (1.9 cm) and possibility of ischemic colitis was kept. On CT angiography, mesenteric vein thrombosis was found, explaining the pain due to mesenteric ischemia. As patient was having thrombotic tendency, along with pancytopenia and haemolytic anemia, we investigated him for paroxysmal nocturnal hemoglobinuria (PNH). On flow cytometry for PNH, 82.3% granulocytes were negative for CD55, 87.4% granulocytes were negative for CD59, and 81.5% of granulocytes were double negative for CD55 and CD59 (PNH clone). So, final diagnosis of PNH was made.
As the patient was severely anaemic, 5 units of packed cells were transfused during his stay at the hospital. His serum haemoglobin level was raised to 10.2 gm%. The patient could not afford to purchase the drug of choice for PNH, eculizumab. He was referred to the department of hematology for bone marrow transplantation, on antithrombotic therapy and danazol.
Discussion
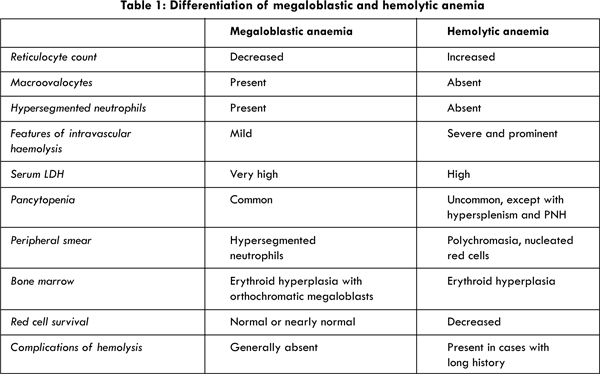
The diagnosis of PNH was delayed in our case as it was not approached systematically and important investigation of reticulocyte count was missed. Moreover, it had overlapping picture with that of megaloblastic anaemia: macrocytosis, pancytopenia and hemolysis. Findings of hemolytic and megaloblastic anaemia are compared in Table 1.
Macrocytosis is defined as a mean cell volume (MCV) of >100 fL and could be due to varied reasons. Depending on presence or absence of megaloblast (large oval shape macrocyte with MCV >125 fL), it is divided into two categories: megaloblastic and non-megaloblastic. Former is due to impaired DNA synthesis and hence, nuclear maturation is delayed due to vitamin B12 and folic acid deficiency. Non-megaloblastic macrocytic anaemia is due to accelerated erythropoiesis and appearance of reticulocytes (hemolytic and post-hemorrhagic anaemia), increased membrane surface area (hepatic disease, obstructive jaundice, splenectomy), spurious macrocytosis (paraproteinemia, inflammation) and alcoholism. Macrocytosis can also be part of hypothyroidism, and serious conditions like myelodysplastic, myelophthisic and aplastic anaemia.
The reticulocyte count provides an initial assessment of the cause of anemia. The normal reticulocyte count by light microscopy is 0.5 to 1.5% of total RBCs. Anaemia with low reticulocyte count usually reflects impaired RBC production, and this can be due to two kinds of defects. Erythropoiesis may be impaired because of a reduction in red cell precursors. Alternatively, red cell production may be ineffective, a condition characterized by erythroid hyperplasia in the bone marrow. To evaluate the causes of anaemia with low reticulocyte counts, analysis of red cell indices is most helpful. Of these, MCV tends to be the most useful index [1]. In cases of anaemia with low reticulocyte count along with macrocytic RBCs, megaloblastic anaemia should be ruled out. In these cases, formation of other blood cells is also affected.
The most characteristic presentation of hemolysis is reticulocytosis with some degree of hyperbilirubinemia due to increased heme catabolism. Other markers are increased serum LDH (direct red cell injury) or low serum haptoglobin, hemoglobinemia, hemoglobinuria, and increased urinary hemosiderin (increased Hb excretion).
The presentation of hemolytic anaemia varies depending on acute or chronic presentation, and hereditary or acquired cause. Patients with congenital hemolytic anemia often remain asymptomatic until late in adult life unless jaundice, crisis or complications of gall bladder disease develop. Acquired hemolytic anemia can develop acutely or gradually over a period of weeks or months. Pallor, icterus or jaundiced complexion may be the first evidence of illness.
The site of RBC destruction is either within circulation (intravascular hemolysis) or within tissue macrophages (extravascular hemolysis). Signs specific for intravascular hemolysis are hemoglobinemia, hemoglobinuria and urinary hemosiderinuria. Normal plasma haemoglobin level is < 1 mg/dL. When it surpasses haptoglobin binding capacity, haemoglobin dimer starts getting excreted in urine, changing colour of urine from faint pink to red and even black (cola beverage coloured). Hemosiderinuria gives reliable evidence of hemoglobinemia in recent past [2]. While hemoglobinuria occurs intermittently in intravascular hemolysis, hemosiderinuria is a constant finding.
Intravascular hemolysis can also be differentiated from extravascular hemolysis by iron profile. While in intravascular hemolysis, there’s iron deficiency and serum ferritin is decreased, in extravascular hemolysis, iron overload occurs and serum ferritin levels are normal or increased.
Specific causes of intravascular hemolysis should be determined by assessing morphological abnormality of RBC in peripheral smear, look for schistocytes, to rule out microangiopathic hemolytic anemia; direct antiglobin or Coomb’s test to rule out autoimmune haemolytic anemia; G6PD assay if disease has developed acutely; flow cytometry to rule out paroxysmal nocturnal hemoglobinuria (PNH). It should be kept in mind that 2-5% of patients of autoimmune haemolytic anaemia can have negative test results [3].
PNH is a rare form of acquired hemolytic anemia. Its pathophysiology lies in the development of immune mediated bone marrow failure and the coexistence of a population of hematopoietic stem cells in which somatic mutation of the X-linked phosphatidylinositol glycan complementation class A (PIG-A) gene results in a partial or absolute deficiency of all proteins normally linked to the cell membrane by a GPI anchor, chiefly CD55 and CD59 [4]. The classical clinical features of PNH are intravascular hemolysis, bone marrow failure and a thrombotic tendency. The definitive diagnosis of PNH must be based on the demonstration that a substantial proportion of the patient’s red cells have an increased susceptibility to complement (C), due to the deficiency on their surface of proteins (particularly CD59 and CD55) that normally protect the red cells from activated C. The gold standard today is flow cytometry, which can be carried out on granulocytes as well as on red cells. A bimodal distribution of cells, with a discrete population that is CD59-, CD55-, is diagnostic of PNH. Usually this population is at least 5% of the total in the case of red cells and at least 20% of the total in the case of granulocytes.
PNH may evolve into aplastic anemia (AA), and rarely (estimated 1–2% of all cases), it may terminate into acute myeloid leukemia. On the other hand, full spontaneous recovery from PNH has been well documented, albeit rarely. More recently, an exceptionally rare form of congenital glycosyl phosphatidyl inositol (GPI) deficiency has been described that involves mutation of an autosomal recessive, phosphatidylinositol glycan complementation class M (pig-m) gene [5]. It is characterised by propensity to venous thrombosis and seizures but not by overt intravascular hemolysis.
To summarise, hemolytic anaemia should be diagnosed early to avoid complications like hepatosplenomegaly, stone formation, kidney injury and infections. All patients of anaemia with raised serum LDH should be evaluated for intravascular hemolysis. If Coomb’s negative, all patients of intravascular hemolysis should be evaluated for PNH. It should be kept in mind that gross hemoglobinuria is present in less than 25% cases of PNH. Cases of PNH are mostly missed and often misdiagnosed as megaloblastic anaemia. This is because both of them have pancytopenia, macrocytosis, and intravascular hemolysis.
Conclusions
In any case of anaemia, the first investigation to approach a case should be the reticulocyte count. Macrocytosis should be worked up keeping both megaloblastic and non megaloblastic causes as differential diagnosis. Macrocytic anaemia with raised serum LDH should not be blindly treated for megaloblastic anaemia, as this is a common finding in hemolytic anaemia. If Coomb’s test negative, the patient of intravascular hemolysis should be evaluated for PNH. Gross hemoglobinuria is present in less than 25% cases of PNH. Megaloblastic anaemia often masquerades PNH. Family history in cases of PNH is important as congenital form of PNH is evolving and has been reported in the literature [5].
References
- Bessman JD, Gilmer PR Jr, Gardner FH. Improved classification of anemias by MCV and RDW. Am J Clin Pathol. 1983;80(3):322–326.
- Fairbanks VF, Gilchrist GS, Brimball B, Jereb JA, Goldston EC. Hemoglobin E trait re-examined: A cause of microcytosis and erythrocytosis. Blood. 1979;53(1):109-115.
- Gilliland BC, Baxter E, Evans RS. Red-cell antibodies in acquired haemolytic anemia with negative antiglobulin serum tests. N Engl J Med. 1971;285(5):252-256.
- Bessler M, Mason PJ, Hillmen P, Miyata T, Yamada N, Takeda N, Luzzatto L, Kinoshita T. Paroxysmal nocturnal hemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO-J. 1994;13:110–117.
- Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosyl phosphatidyl inositol deficiency. Nat Med. 2006;12:846–851.