|
|
|
|
|
Cutaneous Rosai-Dorfman Disease Recurrence in Nasal Region
|
|
|
Kun Chen1, Qian Yang1, Yiyuan Sun1, Runjie Shi1,2
Department of Otorhinolaryngology1, Shanghai Ninth People’s Hospital Affiliated Shanghai Jiao Tong University School of Medicine, Shanghai, China and Otolaryngology2 Institute of Shanghai Jiao Tong University, Shanghai, China. |
|
|
|
|
|
Corresponding Author:
|
Dr. Runjie Shi
Email: runjieshi@hotmail.com
|
|
|
|
|
|
|
|
|
Received:
08-MAY-2014 |
Accepted:
13-JUN-2014 |
Published Online:
15-JUL-2014 |
|
|
|
|
|
|
|
Abstract
|
|
|
|
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a benign non-Langerhans cell histiocytic proliferation which was first described in 1969 by Rosai and Dorfman. The disease most commonly involves the lymph nodes but can be seen in virtually any part of the body. The clinical presentation is usually painless massive bilateral cervical lymphadenopathy. The cutaneous form of Rosai-Dorfman disease (CRDD) is a rare entity that manifests solely with skin papules or nodules and it does not present with the usual myriad of symptoms of classical RDD. Here, we report a case of cutaneous Rosai-Dorfman disease in a 43 year old woman that recurred in the nasal region postoperatively. |
|
|
|
|
|
Keywords :
|
Plasma Cells, Histiocytosis, Skin diseases, Emperipolesis, Anemia.
|
|
|
|
|
|
|
|
|
|
|
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffb4e0050000000803000001000100 6go6ckt5b5idvals|340 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Rosai-Dorfman disease (RDD) is also known as sinus histiocytosis with massive lymphadenopathy (SHML). It is a rare, benign, self-limiting disease of phagocytic histiocytes affecting a young age group presenting with massive bilateral painless cervical lymphadenopathy [1]. The other symptoms may be fever, anemia, elevated erythrocyte sedimentation rate, neutrophilia, and a polyclonal hypergammaglobulinemia [2]. Its etiology is largely obscure. The head and neck region is the preferred site of the extranodal form of RDD. However, purely cutaneous Rosai-Dorfman disease (CRDD) without lymphadenopathy or internal organ involvement rarely occurs [3]. We report a rare, unusual clinical presentation of nasal mass diagnosis as CRDD that recurred in the nasal region postoperatively. We discuss the review of literature and the characteristics of the histopathology and deciding on an appropriate treatment.
Case Report
A 43 year old woman presented with a 2 month history of an initially asymptomatic erythema on the left alar groove, which then developed to a nodule that gradually increased in size (1.0-cm red-yellow size). The microscopic examination of skin lesion done outside revealed a dense dermal infiltrate of histiocytes with abundant lymphocytes, plasma cells. The histiocytes had large vesicular nuclei, abundant nucleoli, and evident foamy cytoplasm. Lymphocytes and plasma cells were obvious within the cytoplasm of these histiocytes, consistent with emperipolesis. Immunohistochemistry revealed that the invading histiocytes were positive for S-100 and CD68 protein and were negative for CD1a. These findings were consistent with the characteristics of cutaneous Rosai-Dorfman disease (CRDD). The patient was treated with subcutaneous injection of hormone for several times, with significant improvement.
After ten months, however, the nodules and papules recurred on the dorsum nasi and gradually enlarged and integrated to form a large plaque. The painless mass gradually increased afterwards, and by the time he came to our hospital, it had grown to about 1x2x2.5 cm in size [Fig.1]. Physical examination revealed it was a non-tender, smooth, erythematous papule. The patient reported no associated symptoms, such as fever, weight loss, or malaise. Lymphadenopathy was not appreciated physical examination. But the computed tomography, performed after the diagnosis of CRDD, revealed several prominent lymph nodes in the neck and submandibular (the maximum was 14x9 mm). As these nodes were deep in the neck and submandibular, biopsy was not performed. All pertinent laboratory results including complete blood count with differential and erythrocyte sedimentation rate were normal. Chest X-ray, bone scan and abdominal ultrasound were unremarkable. The electronic nasopharyngoscope was also performed, but without significant finding.
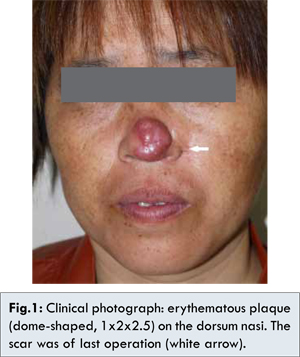
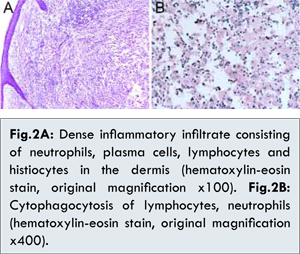
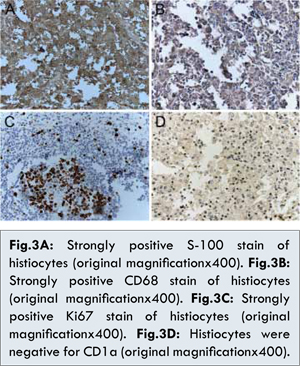
As the mass was large, resection of the mass was performed. The skin specimen revealed a dense dermal infiltrate of histiocytes with abundant lymphocytes, plasma cells and neutrophils [Fig. 2A]. The histiocytes had large nuclei, evident nucleoli, and abundant foamy cytoplasm. Lymphocytes and plasma cells were seen within the cytoplasm of these histiocytes, consistent with emperipolesis [Fig.2B]. Immunohistochemical staining showed that histiocytes were positive for S-100 protein [Fig.3A] and CD68 protein [Fig.3B], but negative for CD1a protein [Fig.3D]. We also performed the immunohistochemical staining for Ki67 protein, and the result was effective, indicating that the histiocytes proliferated obviously [Fig.3C]. All these repeated histopathology and immunohistochemistry in our hospital supported the diagnosis of CRDD. Finally, the lesion was completely resected with skin-grafting. The patient received oral corticosteroid treatment, and the long-term therapeutic efficacy needed to be followed.
Sinus histiocytosis with massive lymphadenopathy (SHML) is a rare disorder that is characterized by prominent painless cervical lymphadenopathy in approximately 90% cases [1,5]. It tends to affect children and young adults [5] and is associated with fever, leukocytosis, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia [1,5].
Although SHML is relatively rare, purely cutaneous RDD (CRDD) is even less common, accounting for approximately 3% of SHML cases in one large study [3]. SHML skin lesions may be diverse and can be found in almost any location, including the face, ears, trunk, extremities or genitalia [5,6]. Several authors have suggested that CRDD is a distinct clinical entity because of its unique epidemiology and the lack of system involvement [6]. Clinically, it can be divided into 3 main types [4]: (i) Papulonodular type: most common (79.5%), and characterized by clustering or satellite papules and/or nodules, up to 1.5 cm in diameter, with red, purple, or brown discoloration; (ii) Indurated plaque type: uncommon (12.8%), and usually presented as plane hyperpigmented plaque with a palpable infiltrated edge; (iii) Tumor type: very rare (7.7%), often presented with red or pinkish dome-shaped, exophytic large mass.
The etiology of CRDD is still unknown. It has been noted to occur sporadically, with occasional clustering, which suggests a genetic or an infectious component. It has been suggested that this disease is related to a reactive disorder since it arises from circulating mononuclear cells and there is an increase in auto-immune antibodies in some affected individuals. Immunological abnormalities are found in a large number of patients and often lead to a clinically unfavorable disease. Epstein-Bar virus and human herpes virus 6 have been proposed as the infectious agent [7]. In our case, the fast recurrence makes us consider the immunological factor.
The diagnosis of CRDD is based on the histologic and immunohistochemical findings of cutaneous lesions. Characteristic features of histology include a dense dermal infiltrate of histiocytes with abundant lymphocytes, plasma cells and neutrophils and the presence of a mixed inflammatory infiltrate. The phenomenon of emperipolesis, a process in which leukocytes transmigrate through the histiocyte, is the key for differentiating CRDD from other diseases. Immunohistochemically, these histiocytes are positive for S-100 and negative for CD1a and either positive or negative for CD68 [4,6]. In our case, we also performed the immunohistochemical staining for Ki67 protein, and the result was effective, indicating that the histiocytes proliferated obviously. Though in many cases, the cutaneous histopathology may be very nonspecific, and the diagnosis may not be clear until there is evaluation of lymph node or other organ involvement, which places the patient at risk for serious complications.
This disease is considered self-limited due to spontaneous resolution and usually does not require aggressive intervention unless leads to a significant functional or physical impairment, such as compression of a vital organ. Different kinds of treatment, such as surgical excision, corticosteroids, thalidomide, cryosurgery, radiotherapy, dapsone, isotretinoin, acitretin, and pulsed dye laser; all of these agents have shown varying rates of success [4,6]. Surgery is indicated as the best choice for dealing with the local disease and it often results in long-term remission [8].
In our case, the inflammatory lesion was localized to the nasal region. Notably, the peculiar manifestation was the dome-shaped, exophytic tumor, which was rarely portrayed in the world literature. The diagnosis of CRDD should be considered in any atypical chronic inflammatory lesion with a histiocytic component involving the skin.
Conclusion
This case report aims at creating knowledge of this rare entity because clinicians, general practitioners and pathologists should always be aware of CRDD in making a differential diagnosis of facial papulonodule.
References
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87(1):63-70.
- Thawerani H, Sanchez RL, Rosai J, Dorfman RF. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol. 1978;114(2):191-197.
- Chuah KL, Tan PH, Hwang SG, Ong BH, Chuan KL. Cutaneous Rosai-Dorfman disease-a pathologic review of 2 cases. Singapore Med J. 2000;41(3):122-125.
- Kong YY, Kong JC, Shi DR, Lu HF, Zhu XZ, Wang J, et al. Cutaneous Rosai-Dorfman disease: a clinical and histopathologic study of 25 cases in China. Am J Surg Pathol. 2007;31(3):341-350.
- Foucar E, Rosai J, Dofman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of entity. Semin Diagn Pathol. 1990;7(1):19-73.
- Wang KH, Chen WY, Liu HN, Huang CC, Lee WR, Hu CH. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154(2):277-286.
- Luppi M, Barozzi P, Garber R, Maiorana A, Bonacorsi G, Artusi T, et al. Expression of human herpesvirus-6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153(3):815-823.
- Gupta K, Bagdi N, Sunitha P, Ghosal N. Isolated intracranial Rosai-Dorfman disease mimicking meningioma in a child: a case report and review of the literature. Br J Radiol. 2011;84(1003): e138-141.
|
|
|
|
|
|
|
Search Google Scholar for
|
|
|
Article Statistics |
|
Chen K, Yang Q, Sun Y, Shi RCutaneous Rosai-Dorfman Disease Recurrence in Nasal Region.JCR 2014;4:249-252 |
|
Chen K, Yang Q, Sun Y, Shi RCutaneous Rosai-Dorfman Disease Recurrence in Nasal Region.JCR [serial online] 2014[cited 2026 May 21];4:249-252. Available from: http://www.casereports.in/articles/4/2/Cutaneous-Rosai-Dorfman-Disease-Recurrence-in-Nasal-Region.html |

|
|
|
|
|