Introduction
Axenfeld-Rieger syndrome (ARS) (MIM# 180500) is a type of neurocristopathy (neural crest maldevelopement) involving three ocular structures-trabecular meshwork (trabeculodysgenesis), iris (iridodysgenesis) and cornea (corneodysgenesis). It is an autosomal dominant disorder with rare occurrence (1 in 2,00,000) and exhibits genetic and morphologic heterogeneity. The underlying genetic defect is known in approximately 60 percent of ARS patients. Majority of studies have shown the involvement of two genes in the causation of ARS, PITX2 and FOXC1that are located on chromosomes 4q25 and 6p25 respectively.
The most common site of chromosomal abnormality is in long arm of chromosome 4. Murray and his associates studied three small families with autosomal dominant Axenfeld-Rieger syndrome and performed linkage analysis in the interval of 4q where the translocations were most common [
1]. Semina and associates identified the affected gene, the paired-like homeodomain transcription factor 2 gene (PITX2) [
2]. In a recently conducted study, it has been shown that the complex distant regulatory matrix within the gene desert located upstream of PITX2 with an essential role in its activity and provides a possible mechanism for ARS in patients [
3]. Nishimura and associates studied two children with congenital glaucoma and systemic abnormalities found translocations involving chromosome 6p25. The cloning of the breakpoint in these children lead to discovery of gene FOXC1 [
4]. A third locus for ARS was mapped to chromosome 13q14 but the gene has not yet been identified [
5].
Case Report
The project was reviewed and approved by Institutional Review board in our institute. All the participants had given consent for participating in the study. All the living members of the family [Fig.1] were interviewed and encouraged for examination. Four members of this family underwent a complete ophthalmologic and systemic evaluation including visual acuity with refraction, slit lamp examination, applanation tonometry, gonioscopy and dilated fundus examination and photography when appropriate. Patients were considered affected if they showed anterior segment dysgenesis with prominent Schwalbe’s line and corectopia with peripheral anterior synechia with systemic features like dermatological, dental and other manifestation. Blood samples were obtained from all the members of the family and were analysed for karyotype in our institute.
Cytogenetic analysis of patient, sibling and parents was done that identified translocation – t(4;11)(q25;pter) in all the affected family members. Both the siblings inherited the chromosomal abberation from their affected mother. This region is known to harbour PITX2 gene, that expalined the clinical presentation in all the affected members.
The family member II1, the mother of affected two children 35 years old, presented to us with both eyes visual acuity of no perception of light (PL), large eyeballs, sclerocornea, scleral thining, glaucoma, microdontia and hypodontia.There was presence of single melanocytic nevus on dorsal aspect of left back. No redundant umbilical fold was found. No other skeletal/cardiac defect was found.
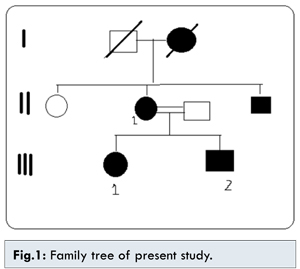
The family member III1 is the daughter of II1 with phthisical right eye with sclerocornea and hypotony. The left eye shows sclerocornea, iris atrophy, pupillary ruff atrophy, polycoria, corectopia [Fig.2] and neovascularisation of iris (temporal 2 clock hours), posterior embryotoxon and 360° peripheral anterior synechiae. Sclera was thin. The vision in left eye with aphakic glasses was 20/80. Corneal diameter was increased. The patient underwent multiple ocular surgeries and is presently aphakic and has trabeculectomy done with qualified success. Systemic examnitations revealed agenesis of unspecified teeth in addition to microdontia and hypodontia. The patient showed a pronounced retrusive lip profile and maxillary hypoplasia with retrognathia. Metric analysis of the jaws showed a highly shortened maxilla (micrognathia). The redundant umbilical fold were found [Fig.3]. No other skeletal/cardiac defect was found. There was presence of single melanocytic nevus on dorsal aspect of left forearm [Fig.4].
The family member III2 son of II1, 10 years old having right eye pthisical with visual acuity no PL. Left eye shows BCVA 20/80, posterior embryotoxon, scleral thinning, iris atrophy, pupil reacting to light, IOP 12 mmHg, cup:disc=0.6:1, NRR=thin, pale. Microdontia, hypodontia were present but redundant umbilical fold was absent. He was operated for congenital heart disease at the age of 5 years. Diagnosis of Axenfeld-Riger syndrome with NVG was made. Left eye trabeculectomy with mitomycin C was done after of intra cameral Avastin injection. On follow-up IOP was 12 mmHg with no medication. Two of the family members showed presence of single large nodulated melanocytic nevus in the back and in forearms of the II1 and III1 respectively.
Discussion
The balanced translocations of chromosome 4 have been reported in various studies, conducted on the patients with ARS. In this present study, the translocation t(4;11)(q25;pter) was seen. In general, ARS patients who display defects in other organ systems, such as teeth or umbilicus, have mutations of the PITX2 gene; meanwhile, in patients with isolated anterior segment dysgenesis, mainly FOXC1 mutations are detected.
It has been observed that the t(4;11)(q25;pter) region harbors PITX2 gene which is involved in the development of eye and craniofacial region. It has recently been reported that the deletions upstream of the PITX2 gene, that removes regulatory elements- CE4 to CE13 leads to ARS in the patients suggesting the presence of a complex distant regulatory matrix upstream of PITX2 with an essential role in its activity and a disruption of which serves as a possible mechanism for the ARS in these patients [
3].
Being a neurocristopathy, Axenfeld-Riger syndrome can involve any cell of neural crest origin. The teeth most frequently reported to be missing are the upper lateral incisors as well as the upper and lower second premolars [
6] which was found in patient, III1 and brother, III2 [Fig.5], however in mother, II1 all the upper incisors were present, but in lower jaws only one lateral incisor was present and all other teeth were absent [Fig.6]. PITX2 has been identified as an activator of the Dlx2 gene that is also expressed in maxillary and mandibular and dental epithelia. Dlx2 is part of the “odontogenichomeobox code” essential for tooth and craniofacial development [
7]. Therefore dental anomalies in ARS patients can be explained by changes in PITX2 gene.
Axenfeld-Rieger syndrome is characterized by complete penetrance, but disease severity shows variability. The same change may result in different manifestations not only in unrelated patients, but also within the same family which was also found in our study in this family. Although both siblings had inherited the same translocation from mother, the disease presentation was variable in these family members [Table1].
Genetic studies like these serves as the early diagnostic indicators of ARS. Cytogenetic investigations can in fact be offered as first amongst the genetic tests series particularly in families where several members are affected, the technique being cost effective and proficient. This will also facilitate inprenatal diagnosis and the family members can be counseled after establishing the genetic basis of ARS.
References
- Murray JC, Bennet SR, Kwitek A. Linkage of Rieger syndrome to the region of epidermal growth factor gene on chromosome 4. Nat Genet 1992;2:46-49.
- Semina EV, Reiter R, Leysens NJ, Alward WLM, Small KW, Siegle-Bartelt J, et al. Cloning and characterization of a novel bicoid- related homeobox transcription factor gene, RGS, involved in Riegersyndrome. Nat Genet. 1996;14:392-399.
- Volkmann BA, Zinkevich NS, Mustonen A, Schilter KF, Bosenko DV, Reis LM, et al. Potential novel mechanism for Axenfeld-Rieger syndrome: deletion of a distant region containing regulatory elements of PITX2. Invest Ophthalmol Vis Sci. 2011; 52(3):1450-1459.
- Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, et al. The forkheadtranscription factor gene FKHL7 is responsible for phenotypes which map to 6p25. Nat Genet. 1998;19:140-147.
- Phillips JC, Del Bono EA, Haines JL, Prale, AM, Cohen, JS, Greff, LJ, et al. A second locus for Rieger Syndrome maps to chromosome 13q14.Am J Hum Genet. 1996;59:613-619.
- Polder BJ, Van’t Hof MA, Van der Linden FP, Kuijpers-Jagtman AM. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dentistry and Oral Epidemiology. 2004;32:217-226.
- Green PD, Hjalt TA, Kirk DE, Sutherland LB, Thomas BL, Sharpe PT, et al. Antagonistic regulation of Dlx2 expression by PITX2 and Msx2: implications for tooth development. Gene Expression. 2001;9(6):265-281.