Introduction
Pyloric atresia is a rare pathology, with an incidence of 1 in 1,00,000 newborns. To date few case reports and small case series exist in the literature [
1,
2,
7]. Due to the rarity of the lesion and because of our patient’s severe and unusual clinical presentation, we chose to report it and discuss the specific laboratory abnormalities, steps leading to the diagnosis, management, and outcome.
Case Report
A 6-day-old female neonate presented to our emergency department due to hypoactivity and poor oral intake (10 ml of milk daily). She also had several episodes of yellowish, non-bilious vomiting and scant meconium. Urine output was not recorded. The baby was very lethargic, sleeping upto eight hours at a time, and often not waking up for feeds. She was seen by a physician on her third day of life and was diagnosed with gastro-esophageal reflux.
Prenatal history revealed a 26-year-old G1P0 mother, who was toxoplasma non-immune, negative hepatitis B surface antigen and had a normal diabetes screen. Pregnancy was uncomplicated, however prenatal ultrasound revealed moderate polyhydramnios and a prominent fetal stomach. Family history was remarkable for parents being first cousins. She was born at 38 weeks of gestation by normal vaginal delivery at a local hospital; her birth weight was 2400 grams and she did not cry for the first 3 minutes, but subsequently improved on flow oxygen. APGAR scores were 6 and 9 at 1 and 5 minutes respectively. She was discharged home on her second day of life.
Examination revealed a lethargic, hypoactive, and hypothermic newborn with a weak cry; blood pressure was 32/17 mm Hg, heart rate 156 beats per minute, respiratory rate 50/min, and oxygen saturation 87% on room air. Weight was 1900 grams, which equals 20% weight loss compared to her birth weight. She had Kussmaul breathing pattern, and showed signs of severe dehydration including sunken eyes and depressed fontanelles, greater than 3-second capillary refill, and tenting and dryness of the skin. Heart and lungs examination showed no abnormality. The abdomen was severely distended, with decreased to absent bowel sounds. She also had occasional staring episodes associated with desaturation.
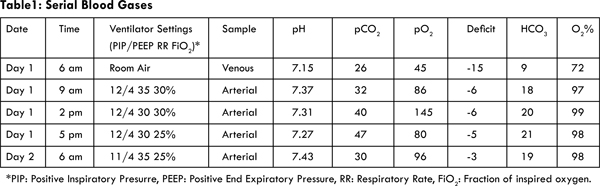
Initial work-up included: blood gases, CBC, electrolytes, total serum protein, uric acid, and liver function tests. Metabolic work-up was also performed. Blood and urine cultures were sent before starting broad-spectrum antibiotics. Urine analysis and urine electrolytes were also assessed. Venous blood gases showed severe metabolic acidosis: pH 7.15, pCO2 26 mmHg, pO2 45 mmHg, base deficit: 15, bicarbonate 9 mEq/L, and O2 72% [Table 1]. Initial blood chemistry was glucose 239 mg/dL, BUN 109 mg/dL, creatinine 7.1 mg/dL, sodium 147 mmol/L, potassium greater than 8.5 mmol/L, chloride 82 mmol/L, carbon dioxide 8 mmol/L, magnesium 3.8 mg/dL, calcium 9.6 mg/dL, and phosphate >20.7 mg/dL, uric acid 34.4 mg/dL, anion gap 57 mmol/L, serum osmolality 350 mOsm/kg and BUN/creatinine ratio was 17.4. Urine analysis revealed a pH of 7, specific gravity of 1.010, protein +2, and glucose of +2. Urine osmolality was 334 mOsm/kg. FENA and urine protein to creatinine ratio were calculated and were 44.6% and 2.2 respectively. CBC, liver function tests, albumin, and initial metabolic work up (ammonia, pyruvate, lactic acid, and serum ketone bodies) showed no significant abnormality. C reactive protein was normal. Chest X-ray was normal. Abdominal film revealed extremely large lucency occupying most of the abdominal cavity likely representing a dilated stomach, with no air seen in the small or large bowel loops [Fig.1].
In summary, initial assessment revealed hypernatremia, hyperkalemia, hypochloremia, high anion gap metabolic acidosis, hypovolemia, and hyperglycemia with evidence of severe renal impairment. This constellation of findings is not characteristic of a specific pathology. Therefore, a broad range of differential diagnoses was considered including infectious, metabolic, renal, endocrinological, and gastrointestinal etiologies.
Sepsis was excluded after negative urine and blood cultures, and rapid improvement after hydration and correction of electrolyte abnormalities. Metabolic disease was ruled out by the normal arterial ammonia, lactate, pyruvate and normal urine organic acid analysis and neonatal screen. The acute kidney injury was secondary to severe dehydration and ensuing renal hypoperfusion. Intrinsic renal disease was excluded by a normal kidney ultrasound, and renal function normalized by sixth day of intravenous fluid therapy.
Neonatal diabetes was also considered briefly due to the hyperglycemia, acidosis, and dehydration. In the first two days of hospitalization insulin drip was required to control glucose level, after which hyperglycemia resolved without recurrence. Congenital adrenal hyperplasia could explain the shock-like presentation, hyperkalemia, and hypovolemia; but did not explain the hyperglycemia and lack of ambiguous genitalia. The history of non-bilious vomiting and polyhydramnios coupled with the abdominal distension on physical exam, hypochloremia, and findings on abdominal film (‘single bubble sign’); were highly suspicious of gastric outlet obstruction. Despite the unusual finding of metabolic acidosis, a gastrointestinal etiology became the most likely differential.
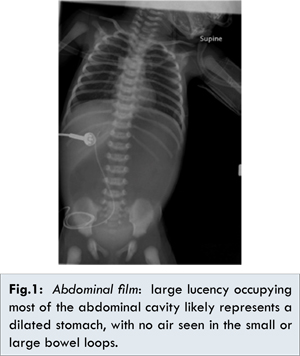
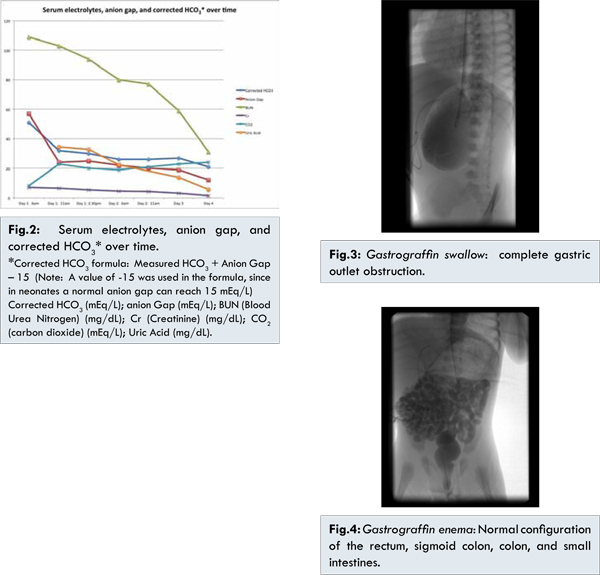
In the emergency room central access was obtained, and intravenous fluid resuscitation (20 ml/kg normal saline pushes) was administered. She was intubated to secure the airway due to the respiratory distress and severe metabolic acidosis. Nasogastric decompression was performed revealing blood-tinged milk, and she was transferred to the neonatal intensive care unit for further management. With progressive hydration serial arterial blood gases showed improvement in the metabolic acidosis [Table 1]. Similar improvements were seen in the electrolyte abnormalities, anion gap, and corrected bicarbonate [Fig.2]. Corrected bicarbonate is a calculation performed in the setting of anion gap metabolic acidosis to reveal an accompanying non-gap metabolic acidosis or metabolic alkalosis. If the corrected bicarbonate is above normal, then a concomitant primary metabolic alkalosis is present, as was the case seen with our patient. After stabilization, and on the fifth day of hospitalization, she underwent a gastrograffin swallow, which revealed complete gastric outlet obstruction [Fig.3]. A gastrograffin enema showed normal configuration of the rectum, sigmoid colon, colon, and small intestines [Fig.4]. On the sixth day of hospitalization, exploratory laparotomy was performed and she was found to have a completely obstructed pylorus, with complete partition of the pyloric canal and thickened mucosal and muscularis diaphragm consistent with type 2 pyloric atresia. Gastroduodenostomy was performed without complications.
The post-operative course was relatively smooth. Feeding was initiated on the 4th post-operative day, and full enteral feedings were reached within two weeks. By that time she was 28 days of age, and had reached a weight of 2350 grams. Currently she is a healthy 2 years 3 months old child who is thriving with normal developmental milestones.
Discussion
Our patient’s late presentation resulted in the unusual finding of high anion gap metabolic acidosis. The etiologies of high anion gap metabolic acidosis are well documented in the literature, and include lactic acidosis, ketoacidosis, renal failure, toxin ingestion, and organic acidemia secondary to metabolic disorders. The expected metabolic alkalosis associated with pernicious vomiting due to her pyloric atresia was masked by the severe acute kidney injury resulting from extreme dehydration, hypovolemia and renal hypoperfusion. This acute uremia was the main cause of her severe anion gap metabolic acidosis. Lactate level was slightly elevated but is unlikely to have contributed to the gap acidosis.
Congenital anomalies of the gastrointestinal tract a relatively common, however congenital pyloric atresia is a rare anomaly accounting for less that 1% of all intestinal atresias [
8]. The incidence is equal among male and female neonates [
1]. There are three anatomic variants of pyloric atresia; Type 1 the pyloric membrane or web is the most common accounting for 57% of the cases. In Type 2 (34%) a fibrous solid cord replaces the pyloric canal, and in Type 3 (9%) there is a complete disconnection between the stomach and duodenum [
9]. Our case index had a type 2 lesion.
Patients usually present soon after birth due to non-bilious vomiting [2,10], which is the most common clinical feature at presentation (100% of cases) in addition to epigastrium distension (68% of cases) [
5]. In a case series by Llce et al., the condition was often associated with a low birth weight [
1]. Prenatally the diagnosis is often suspected due to polyhydramnios (63% of cases) [
5]. Fetal ultrasound also often shows a dilated stomach and at times fluid distension of esophagus [
6,
8,
9]. Postnatal diagnosis also relies on imaging, with abdominal radiograph showing the characteristic ‘single-bubble’ sign in up to 98% of the cases [
5]. Postnatal ultrasound can also be performed and may show an elongated and echogenic pylorus with no patent channel [
2,
6].
For all types of pyloric atresia the management is surgical. In Type 1, excision of the pyloric web is performed followed by a Heineke-Mikulicz pyloroplasty; whereas Type 2 and 3 are treated by gastroduodenostomy [
8]. Our patient’s Type 2 lesion was corrected as recommended by gastroduodenostomy. Pyloric atresia can occur as an isolated lesion, or can be associated with other anomalies in approximately 33%-55% of cases [
1,
6,
10]. The most commonly associated abnormality is epidermolysis bullosa [
7,
10,
11]. Isolated congenital pyloric atresia has an excellent prognosis with appropriate surgical management, however the presence of associated anomalies negatively impact morbidity and mortality [6].
References
- Ilce Z, Erdogan E, Kara C, Celayir S, Sarimurat N, Senyüz O, et al. Pyloric atresia: 15-year review from a single institution. J Pediatr Surg. 2003;38(11):1581-1584.
- Okoye B, Parikh D, Buick R, Lander A. Pyloric atresia: Five new cases, a new association, and a review of the literature with guidelines. J of Pediatr Surg. 2000;35(8):1242-1245.
- Muller M, Morger R, Engert J. Pyloric atresia: a report of four cases and review of literature. Pediatr Surg Int. 1990;5:276-279.
- Andriessen MJ, Matthyssens LE, Heij HA. Pyloric atresia. J of Pediatr Surg. 2010;45(12):2470-2472.
- Parshotam G, Ahmed S, Gollow I. Single or double bubble: Sign of trouble! Congenital pyloric atresia: Report of two cases and review of literature. J Paediatr Child Health. 2007;43(6):502-503.
- Merrow AC, Frischer JS, Lucky AW. Pyloric atresia with epidermolysis bullosa: fetal MRI diagnosis with postnatal correlation. Pediatr Radiol. 2013;43(12):1656-1661.
- Al-Salem A. Congenital pyloric atresia and associated anomalies. Pediatr Surg Int. 2007;23(6):559-563.
- Al-Salem A, Abdulla M, Kothari M, Naga M. Congenital pyloric atresia, presentation, management, and outcome: A report of 20 cases. J Pediatr Surg. 2014;49(7):1078-1082.
- Joshi P, Mundada D, Shetty S, Oak S, Parelkar S, Kapadnis S, et al. Pyloric atresia: Three cases and review of literature. Afr J Paediatr Surg. 2014;11(4):362-365.
- Ksia A, Zitouni H, Zrig A, Laamiri R, Chioukh F, Ayari E, et al. Pyloric atresia: A report of ten patients. Afr J Paediatr Surg. 2013;10(2):192-194.
- Farmakis SG, Herman TE, Siegel MJ. Congenital pyloric atresia, type B; with junctional epidermolysis bullosa. J of Perinatol. 2014;34(7):572-573.