|
|
|
|
|
Denys Drash Syndrome: A Rare Presentation
|
|
|
Sandeep Gupta, Arun Kumar Maurya, Kumar Rajiv Ranjan, Dilip Kumar Pal
Department of Urology, Institute of Post-Graduate Medical Education and Research and S.S.K.M. Hospital, Kolkata-700020, West-Bengal, India. |
|
|
|
|
|
Corresponding Author:
|
Dr. Sandeep Gupta
Email: urologyipgmer@gmail.com
|
|
|
|
|
|
|
|
|
Received:
27-APR-2016 |
Accepted:
04-JUL-2016 |
Published Online:
15-SEP-2016 |
|
|
|
|
|
|
|
Abstract
|
|
|
|
A 30 month old phenotypically female child presented to our outdoor with painless progressive lump in left upper abdomen with ambiguous genitalia. Ultrasound and CT findings were suggestive of malignant left renal mass with right normal and left streak gonad in abdomen. Karyotyping shows 46XY. Patient had persistent proteinuria and hypertension. Histopathology report of left renal mass was biphasic Wilms tumor with favorable histology and mesangial sclerosis in the non-malignant part. Patient had a classic triad of Denys Drash syndrome. In most of the reported cases, patient presented with end stage renal disease but in our case overt feature of nephrotic syndrome or end stage renal disease were not associated, which is a rare phenomenon. |
|
|
|
|
|
Keywords :
|
Denys-Drash syndrome, Gonads, Hypertension, Karyotyping, Kidney Failure, Nephrotic Syndrome.
|
|
|
|
|
|
|
|
|
|
|
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff645810000000e904000001001000 6go6ckt5b5idvals|661 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Denys-Drash syndrome (DDS) is mainly group of three symptoms: nephrotic syndrome due to diffuse mesangial sclerosis, male pseudohermaphroditism, and high probability to develop Wilms tumor and gonadoblastomas [ 1, 2]. Among three symptoms nephropathy is the constant one. The syndrome is caused by dominant mutations in the WT1 gene [ 3] located on chromosome 11p13 that encodes a zinc finger DNA binding protein, Wilm’s tumor suppressor 1 (WT1) [ 4]. WT1 plays an important role in the development of several organs, including the kidney [ 5] and the urogenital tract [ 6, 7].
Case Report
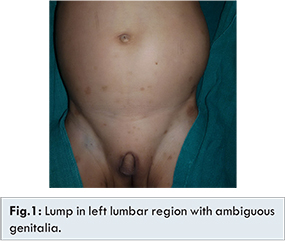
A 30-month-old child presented with painless progressive enlargement of left side of the abdomen from last three months with a history of under development of external genitalia since birth. He has an elder sister of 7 years having no similar complaints or co-morbidities. At admission, the child had a weight of 10 kg (<50th centile) and length 82 cms (<50th centile). The blood pressure was 130/90 mmHg. A large lump was palpable in the left lumbar region, involving almost left half of the abdomen without crossing the midline. The external genitalia were ambiguous with a stretched penile length of 1.5 cm [Fig.1]. Labioscrotal folds were present and bilateral gonads were not palpable. There was a single urogenital opening along with penoscrotal hypospadias. There were some hyper-pigmented patches over the lower part of abdomen which measures from five mm to two cm in diameter. The rest of the physical examination and development were normal.
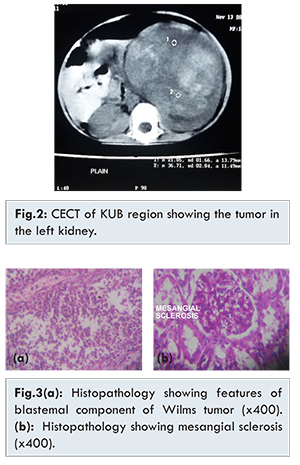
On investigation, haemoglobin was 6.7 g/dL and TLC 20,800/mm3. She had normal renal biochemical parameter and serum electrolytes. Total serum proteins were 7.1 g/dL (albumin 4.5 and globulin 2.6). Urinalysis showed 2+ proteinuria, 5-8 WBCs per high power field and 12-15 dysmorphic red cells/HPF. Serum cortisol (16.69 µg/dL morning) and 17 alpha-OH progesterone (1.76 ng/mL) levels were in the normal range. Serum testosterone (<20 ng/mL levels) were low for prepubertal range. The peripheral blood lymphocyte karyotype was 46, XY. Chest X-ray and liver function test were normal. Ultrasound of the abdomen showed a left renal mass. No Wolfian duct structures were seen in the pelvis. CT of the abdomen showed a large patchy enhancing, heterogeneous SOL arising from left kidney with a normal right kidney [Fig.2]. The right gonad was seen separately from the tumor mass. Uterus was infantile and left gonad was found to be streaked. Left radical nephrectomy was done. The mass was free from the surrounding structures. Histologic evaluation of the tumor mass was compatible with a diagnosis of favourable biphasic Wilms’ tumor and margins were free from tumor [Fig.3(A)]. Renal mesangial sclerosis was found [Fig.3(B)] from the tumor free areas. Patient is on regular follow up and till one year no clinical feature of nephrotic syndrome seen. Postoperative chemotherapy has been started as per protocol.
In Denys Drash syndrome, nephropathy is characterized by onset of proteinuria between birth to two years of age, although it may develop as late as 6 to 14 years of age [ 9]. It is important to consider the diagnosis of Denys-Drash syndrome in any patient with unexplained nephropathy, particularly young phenotypic girls and children with ambiguous genitalia, or those presenting with an early Wilms’ tumor. Nephropathy is a constant feature of DDS. Renal dysfunction usually progresses to end stage renal disease by the age of three years. In incomplete forms of the syndrome, nephropathy exists either with Wilms’ tumor or with intersex disorder. In our case, overt nephrotic syndrome has not developed yet till 2.5 years of age. Genital abnormalities are frequently seen in patients with 46 XY karyotype, while those with 46 XX show less or no genital abnormality. In patients with ambiguous genitalia and renal disease a close follow-up should be maintained, with regular renal ultrasound scans, to look for development of Wilms’ tumor and gonadal malignancy. Conversely, in patients with Wilms’ tumor and intersex disorder, renal function should be serially assessed to document development of proteinuria and renal insufficiency.
Conclusions
The Denys-Drash syndrome should be suspected in children with XY gonadal dysgenesis. The proteinuria should be sought in such children, and if present, consultation with genetic and nephrology specialists is warranted, these patients are at high risk of developing bilateral Wilms tumor, ultrasound screening is recommended. Dialysis and ultimately renal transplantation is the treatment for these patients who are suffering from end stage renal disease.
References
- Denys P, Malvaux P, Van Den Berghe H, Tanghe W, Proesmans W. Association of an anatomo-pathological syndrome of male pseudohermaphroditism, Wilms’ tumor, parenchymatous nephropathy and XX/XY mosaicism. Arch Fr Pediatr. 1967;24:729-739
- Drash A, Sherman F, Hartmann WH, Blizzard RM. A syndrome of pseudo-hermaphroditism, Wilms’ tumor, hypertension, anddegenerative renal disease. J Pediatr. 1970;76:585-593.
- Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys Drash syndrome. Cell. 1991;67:437-447.
- Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509-520.
- Davies JA, Ladomery M, Hohenstein P, Michael L, Shafe A, Lee Spraggon, et al. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet. 2004;13:235-246.
- Hohenstein P, Hastie ND. The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet. 2006;15:196-201.
- Roberts SG. Transcriptional regulation by WT1 in development. Curr Opin Genet Dev. 2005;15:542-547.
- Wagner N1, Wagner KD, Theres H, Englert C, Schedl A, Scholz H. Coronary vessel development requires activation of the TrkBneurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 2005;19:2631-2642.
- Eddy AA, Mauer SM. Pseudohermaphroditism, glomerulopathy, and Wilms’ tumor (Drash syndrome): frequency in end-stage renal failure. J Pediatr. 1985;106:584-587.
|
|
|
|
|
|
|
Search Google Scholar for
|
|
|
Article Statistics |
|
Gupta S, Maurya AK, Rajiv Ranjan K, Pal DKDenys Drash Syndrome: A Rare Presentation.JCR 2016;6:432-434 |
|
Gupta S, Maurya AK, Rajiv Ranjan K, Pal DKDenys Drash Syndrome: A Rare Presentation.JCR [serial online] 2016[cited 2026 Jun 25];6:432-434. Available from: https://www.casereports.in/articles/6/3/Denys-Drash-Syndrome.html |

|
|
|
|
|