Introduction
Carnitine palmitoyl transferase 1 (CPT1) deficiency is a rare autosomal recessive fatty acid oxidation defect. The carnitine palmitoyl transferase (CPT) system carries out both enzyme and transporter functions. It consists of CPT1 and CPT2. CPT1 enzyme is located within the external mitochondrial membrane and whose function is to conjugate long chain fatty acids to carnitine. Carnitine is required for transfer of long chain fatty acid from the cytoplasm to the mitochondria for oxidation [
1]. The reaction leads to the transfer of the acyl group to carnitine to form acyl carnitine. CPT2 is an inner mitochondrial protein. Its function is to reverse the CPT1 reaction, regenerating acyl-CoA in the matrix and releasing carnitine [
2]. There are three isoforms of CPT1. CPT1A is liver isoform, CPT1B is muscle isoform and CPT1C is brain isoform. Our case fits in with CPT1A subtype. The gene is localised in 11q13.1-q13.2. CPT1A is expressed in liver, kidney, leukocytes and fibroblasts [
3].
Case Report
A 14 year old boy was admitted in paediatric intensive care unit for fever, decreased food intake, lethargy and altered sensorium for two days. There was no history of trauma, headache, visual disturbances, vomiting, seizures, involuntary movements or drug intake. His past history at three years of age was suggestive of brief drowsiness which improved after treatment. He was a term baby born to consanguineous parents by Caesarean section with a birth weight of 2.75 kg. There was no history of unexpected death or similar illness, in siblings or in the broader family members. Prior to hospitalisation, his scholastic performance was good.
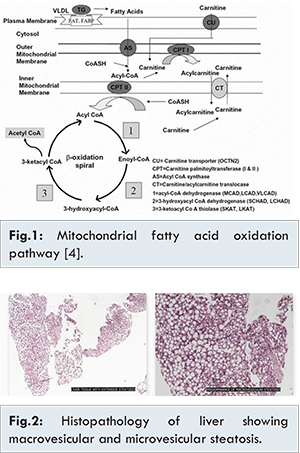
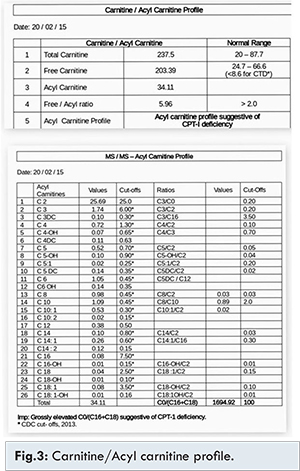
His Glasgow coma scale was 9/15 at presentation. Clinical examination revealed hepatomegaly and liver was palpable 12 cm below right costal margin. During the first week of hospital stay, he developed icterus, pedal edema and ascites. In addition, he developed dystonic movements of limbs. Initial investigations revealed high anion gap metabolic acidosis, hypoglycaemia and hyper-ammonemia. Urine ketones were negative. Blood ketones were normal. Full blood count was normal. Serial liver function tests were done. During critical period it showed hyperbilirubinaemia (direct bilirubin: 21.8 mg/dL; indirect bilirubin: 10.8 mg/dL), raised liver enzymes (SGPT: 309 U/L, SGOT: 283 U/L, GGT: 116 U/L), normal coagulation profile and serum proteins. Ultrasound abdomen revealed hepatomegaly and ascites. Serum ceruloplasmin and urine copper were normal. Slit lamp examination showed no Kayser-Fleischer ring. Viral markers for hepatitis A, B, C and E were negative. Coomb’s test and anti-nuclear antibodies were negative. MRI brain, CSF analysis, echocardiogram and serum creatinine were normal. Blood and CSF cultures were sterile. Liver biopsy showed diffuse microvesicular and macrovesicular steatosis [Fig.2]. Extended metabolic screening showed high total carnitine, high free carnitine, normal free/acyl carnitine ratio and grossly elevated C0/(C16+C18) suggestive of CPT-1 deficiency [Fig.3].
He was ventilated with volume control mode. He was commenced on intravenous fluids and intravenous antibiotics according to our hospital standard protocol. Cerebral edema protective measures were followed including intravenous mannitol and 3% saline. In addition, liver protective measures were carried out. He received partial parenteral nutrition consisting of carbohydrate and protein. Five cycles of sustained low efficiency dialysis was done for hyper-ammonemia and liver protective measures were continued. Tracheostomy was done after weaning off from ventilator. As his sensorium improved, tracheostomy closure was done. Serial investigations showed decreasing trend of ammonia, bilirubin levels and liver enzymes. Feeds were replaced for parenteral nutrition. It was given through nasogastric tube, before changing to oral feeds. The feeds composed of high carbohydrate, normal protein and low fat. He remained euglycemic and hemodynamically stable. Follow-up showed normal liver function, serum ammonia levels and he was continued on planned diet.
Discussion
CPT1A deficiency is very rare and only 40 cases are reported so for [
5]. CPT1 deficiency usually presents in infancy. The child would present with altered sensorium, hepatomegaly, non-ketotic hypoglycemia, increased plasma carnitine levels and mild hyperammonemia. It is triggered by fasting or viral illness [
5]. Metabolic work up would show elevation of free and short-chain acylcarnitine, with low levels of long-chain acylcarnitine. Measurement of total C0 or the ratio of free carnitine to long-chain species (C16 and C18) has been used in making a diagnosis of CPT1A deficiency. It is confirmed by CPT1 assay in fibroblasts, in which the CPT1 activity would be reduced [
5]. Diagnosis is confirmed by doing genetic testing. Death as a result of fulminant hepatic failure had happened in a 17 year old, even though fatty acid oxidation defect had been detected earlier [
6]. CPT1 deficiency is included in newborn screening programs in many countries to detect presymptomatic patients and initiate early treatment [
7].
Our patient was unique because he had presented and diagnosed during adolescence, as many cases were not reported in this age group. He had presented with metabolic encephalopathy along with liver involvement. Neurologically he had dystonia of both upper and lower limbs, which had improved completely. The maximum bilirubin went up to 32.6 mg% (547 mmol/L), which had fully resolved. His three month follow up liver function test was within normal range along with decrease in size of the liver. Resolution of such severe liver involvement is not reported in the literature.
One should avoid hypoglycaemia especially during period of illness or surgery. It can be done with intravenous fluids like 10% dextrose or emergency regimens to prevent hypoglycaemic attacks. High carbohydrate and low fat diet, especially medium chain triglycerides should be advised. Liver function test should be done during the period of illness [
5].
Conclusion
Fatty acid oxidation defect should be considered in a child who presents with hepatic encephalopathy during fasting or febrile illness. Emergency treatment protocol should be followed in case of crisis. Low fat and high carbohydrate diet should be strictly followed in children who are diagnosed with CPT1 deficiency.
References
- Roe CDJ. Carnitine palmitoyl transferase deficiency. In: Scriver C BA, Sly W, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th edn. New York: McGraw-Hill; 2001, 2297-2326.
- McGarry JD, Brown NF. The mitochondrial carnitine palmitoyl transferase system. From concept to molecular analysis. Eur J Biochem.1997;244:1-14.
- Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet Semin. 2006;15:77-85.
- Shekhawat P, Bennett MJ, Sadovsky Y, Nelson DM, Rakheja D, Strauss AW. Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am J Physiol Endocrinol Metab. 2003;284:1098-1105.
- Bennett M, Santani A. Carnitine Palmitoyltransferase 1A Deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al. editors. Gene reviews. Seattle (WA): University of Washington, Seattle; 1993-2016.
- Brown NF, Mullur RS, Subramanian I, Esser V, Bennett MJ, Saudubray JM, et al. Molecular characterization of L-CPT I deficiency in six patients: insights into function of the native enzyme. J Lipid Res. 2001;42:1134-1142.
- Borch L, Lund AM, Wibrand F, Christensen E, Sondergaard C, Gahrn B, et al. Normal levels of plasma free carnitine and acylcarnitines in follow-up samples from a presymptomatic case of Carnitine palmitoyl transferase 1 (CPT1), Deficiency detected through newborn screening in Denmark. JIMD report. 2012;3:11-15.