Introduction
Kikuchi-Fujimoto disease (KFD) is an enigmatic disease of uncertain origin. It presents in women aged about 40 years of Asian descent with tender cervical lymphadenopathy accompanied by fever [
1]. There is no laboratory test which can specifically diagnose this entity. Fine needle aspiration cytology only reveals features of necrotizing lymphadenitis. Histopathological examination shows a paracortical necrotic lesion with presence of karyorrhectic debris and proliferation of numerous histiocytes, large transformed lymphocytes and CD8 lymphocytes. The diagnosis is made by exclusion of sinister mimics such as lymphomas, lupus and infections [
1]. We discuss a case of KFD presenting in a 34 year old lady after taking due consent.
Case Report
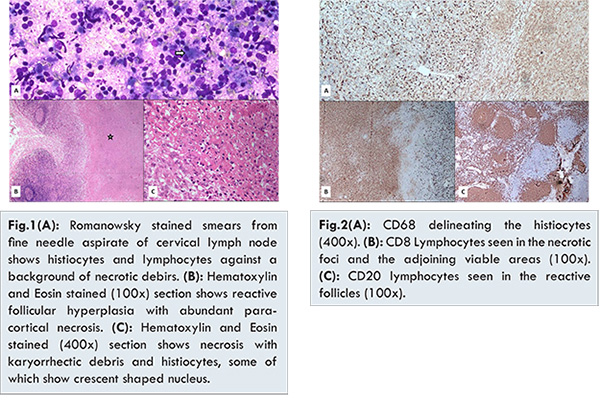
A 24 year old lady reported to the Out Patient Department with complaints of low grade fever and throat pain of 10 days duration. On examination she had enlarged tender bilateral cervical lymph nodes largest measuring 2x1.5x1.5 cm. She had no history of night sweats. There was no hepatosplenomegaly. Investigations revealed a raised ESR, leucopenia (3600/mm3) and few activated lymphocytes on peripheral blood smear. Romanowsky stained fine needle aspirate smears were paucicellular and composed of necrotic debris with scattered histiocytes and mature lymphocytes [Fig.1A]. Ziehl-Neilson stained smears did not reveal presence of acid fast bacilli. An opinion of necrotizing lymphadenitis was offered. The lymph node biopsy was received with specimen measuring 2x1.5x1 cm. The cut surface showed whitish necrotic areas. Hematoxylin and Eosin stained sections revealed partially effaced lymph node architecture with extensive necrosis of the paracortical areas and abundant karyorrhectic debris [Fig.1B,1C]. The necrotic areas were bordered by numerous histiocytes [Fig.1C], some of which had characteristic crescent shaped nucleus. Accompanying these histiocytes were few large transformed lymphocytes. No Reed Sternberg cells, eosinophils, neutrophils, plasma cells or epithelioid cell granulomas were seen. Immunohistochemistry work-up delineated the presence of the numerous CD68 positive histiocytes [Fig.2A]. The karyorrhectic debris and the surrounding areas showed presence of numerous CD8 positive T cells [Fig.2B]. The CD20 positive B cells were restricted to the reactive follicles [Fig.2C]. An opinion of KFD was offered. The subsequent tests for ANA and dsDNA were negative. The patient was put on conservative management with antipyretics and analgesics. At the end of one week of treatment the patient become afebrile. At the end of two months the lymphadenopathy resolved.
Kikuchi-Fugimito was initially reported in Japanese women by Kikuchi and Fugimoto et al. in the year 1972 [
2,
3]. It has been commonly described in the Asian women, though other ethnic populations are also affected [
1]. The patient has an acute or sub-acute presentation of tender cervical lymphadenopathy up to 4 cm, accompanied by fever. The fever is of low grade and lasts for 1 week with rare cases lasting upto one month. The febrile episodes are associated with upper respiratory symptoms [
1,
4]. Generalized lymphadenopathy and hepatosplenomegaly are rarely seen [
1,
4,
5]. Cutaneous manifestations are commonly documented ranging from erythematous papules to ulcers over the region of face and upper body [
6]. Weight loss, night sweats and systemic symptoms are seen in exceptional cases [
4,
5]. Though laboratory findings lack uniformity, elevation of lactate dehydrogenase, transaminases and erythrocyte sedimentation rate have been reported in many cases [
1,
5]. About 50% of cases have leucopenia and about 25% of cases show presence of atypical lymphocytes in the peripheral blood [
1,
4]. Fine needle aspiration cytology features include presence of necrotic debris, histiocytes and karyorrhectic debris. The accuracy rate is about 56% which warrants a biopsy for definitive diagnosis [
7]. Histopathology of the involved lymph nodes shows a partially effaced architecture with a circumscribed area of necrosis with abundant karyorrhectic debris. The necrotic edge is populated by numerous histiocytes which have a bland morphology with few having a crescent shaped nuclear morphology. Clustering of plasmacytoid dendritic cells at the margins of the necrotic areas have also been reported in many cases. Few cases also show foci of proliferation of numerous large transformed lymphocytes admixed with tingible body macrophages. Some cases show evidence of thrombosed vessels. Characteristically absent are neutrophils and eosinophils. The preserved nodal areas usually show reactive follicular hyperplasia [
1]. Kuo et al. had proposed classification based on the evolving histologic patterns: proliferative, necrotizing and xanthomatous [
5]. However this hypothesis is yet to be proved due to the lack of serial biopsies in patients. The importance of immunohistochemistry is in ruling out a possibility of lymphoma especially in cases having large transformed lymphocytes. Histiocytes show positivity for CD68. The admixed lymphocytes are of CD8 positive in contrast to lymphomas having a paracortical expansion which are CD4 positive [
1,
4,
8]. Flow cytometry has a role in differentiating Kikuchi-Fujimoto disease from other reactive lymphadenopathies and lymphoma [
1]. However there is no role of molecular studies in making a diagnosis of Kikuchi-Fujimoto disease [
9]. The pathogenesis still remains uncertain. However, based on the histopathology and clinical presentation, the role of a viral etiology is the most accepted hypothesis [
9]. The features which make the viral etiology probable are - prodromal illness of upper respiratory tract, atypical lymphocytes on peripheral blood smear, paracortical expansion of lymph node with proliferation of large transformed lymphocytes and lack of response to antibiotics. The unique ethnic distribution makes a probable genetic predisposition tangible; nevertheless no specific genetic profile has been identified [
9]. In light of the clinical presentation, laboratory features and histopathological patterns, it can be hypothesized that Kikuchi-Fugimoto disease is because of a hyperacute T-cell mediated cytotoxic injury triggered by a viral antigen in a genetically susceptible individual [
1].
The primary differential diagnosis include: lymphomas, SLE and infections. Lymphomas can present with necrosis of the lymph node. Hodgkin’s lymphoma can be distinguished based on the presence of Reed Sternberg cells, neutrophils and eosinophils. Anaplastic large cell lymphoma cells have CD30 phenotype which is absent in histiocytes and plasmcytoid dendritic cells. Lack of B cell immunophenotype rules out the diagnosis of B-cell lymphoma. Peripheral T-cell lymphoma presents with a para-cortical expansion, but have a CD4 immunophenotype in contrast to a predominant CD8 lymphocytes in KFD [
1]. SLE remains the most exigent differential diagnosis. In addition to the necrosis and paracortical expansion, lupus lymphadenitis shows presence of hematoxylin bodies, Azzopardi phenomenon, abundant plasma cells and neutrophils [
10]. Lack of the additional histopathological features, careful clinical examination for systemic manifestations and absence of auto-antibodies such as anti-nuclear antibody and ds-DNA helps to differentiate KFD from SLE lymphadenitis. Many infectious etiologies can present with paracortical expansion, necrosis and histiocytic proliferation. The viral etiologies usually have predominant CD4 lymphocytes accompanied by plasma cells and neutrophils. Tuberculosis, histoplasmosis, cat-scratch disease and leprosy present as necrotizing granulomatous lymphadenitis with presence of epithelioid cells, giant cells and granulomas [
11]. KFD is a self-limiting disease with a recurrence rate of 3% [
11]. The disease usually resolves by one month. Due to the uncertain etiology, the management is directed towards alleviating the symptoms using conservative means by the use of antipyretics and analgesics [
1,
11].
Conclusion
Kikuchi-Fujimoto disease is a unique entity which needs to be identified in patients because inspite of having a sinister presentation of enlarged tender lymph nodes and abundant necrosis on histomorphology, the condition is self-limiting and requires basic symptomatic management. However, use of histopathology, immunohistochemistry and ancillary serological tests should be used to rule out the possibility of lymphoma, SLE and infections.
Acknowledgement
Brig Reena Bharadwaj and Col. Kavita Sahai for review of the histopathology slides.
References
- Hutchinson CB, Wang E. Kikuchi-Fujimoto Disease. Arch Pathol Lab Med. 2010;134:289-293.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:378-380.
- Fujimoto Y, Kozima Y, Hamaguchi K. Cervical necrotizing lymphadenitis: a new clinicopathological agent. Naika. 1972;20:920-927.
- Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Kuo T. Kikuchi’s disease (histiocytic necrotizing lymphadenitis): a clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology, and DNA ploidy. Am J Surg Pathol. 1995;19:798-809.
- Yasukawa K, Matsumara T, Sato-Matsumara KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Tong TR, Chan OW, Lee KC. Diagnosing Kikuchi disease on fine needle aspiration biopsy: a retrospective study of 44 cases diagnosed by cytology and 8 by histopathology. Acta Cytol. 2001;45:953-957.
- Hassan M, Anees A, Zaheer S. Kikuchi-Fujimoto Disease: Diagnostic Dilemma and the Role of Immunohistochemistry. J Clin Med Res. 2009;1:244-246.
- Deaver D, Horna P, Cualing H, Sokol L. Pathogenesis, diagnosis, and management of Kikuchi-Fujimoto disease. Cancer Control. 2014;21:313-321.
- Hsiang-Cheng, Jenn-Huang CL, Guo-Shu H, et al. Systemic lupus erythematosus with simultaneous onset of Kikuchi-Fujimoto’s disease complicated with antiphospholipid syndrome: a case report and review of the literature. Rheumatol Int. 2005;25:303-306
- Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol. 2004;122:141-152.