|
|
|
|
|
Acrocephalosyndactyly Syndrome: Challenging Diagnosis
|
|
|
laws on abortion abortion clinics in memphis tn bistromc.org Ada Paloma Soto Brambila1,2, Paloma Rivero Moragrega3, Ingrid Patricia Dávalos Rodríguez1,2, Juan Francisco Santoscoy Gutiérrez3 1División de Genética and 3División de Neurociencias, Centro de Investigación Biomédica de Occidente, Instituto Mexicano del Seguro Social. Guadalajara, Mexico; 2Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara. Guadalajara, Mexico. |
|
|
|
|
|
Corresponding Author:
|
|
Dr. Juan Francisco Santoscoy Gutiérrez Email: f.santoscoygtz@gmail.com |
|
|
|
|
|
|
|
|
Received:
21-FEB-2017 |
Accepted:
27-APR-2017 |
Published Online:
30-MAY-2017 |
|
|
|
|
|
|
|
Abstract
|
|
|
|
Introduction: Acrocephalosyndactyly syndromes are a specific group of the syndromic craniosynostosis characterized by cranial and limb abnormalities. There have been described different phenotypic variations that are classified as individual syndromes, these include: Saethre-Chotzen, Crouzon, Baller-Gerold, Apert, and Pfeiffer. Case Report: This case presents a 29-year-old male with the classic Saethre-Chotzen syndrome phenotype: bilateral coronal synostosis without intellectual disability, bilateral ptosis, low frontal hairline, small ears with prominent crus, and cutaneous syndactyly. Conclusion: To distinguish Saethre-Chotzen syndrome from its differential diagnosis it is essential to perform an adequate clinical evaluation and a comparison of the patient’s characteristics with the phenotypic features specific for each acrocephalosyndactyly syndrome. Molecular analysis can orientate towards the diagnosis, but the physical exam is still the primary diagnostic tool. |
|
|
|
|
|
Keywords :
|
Acrocephalosyndactylia, Congenital Microtia, Craniosynostoses, Skull, Syndrome.
|
|
|
|
|
|
|
|
|
|
|
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff288019000000af03000001000c00 6go6ckt5b5idvals|760 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Craniosynostosis is defined as the premature fusion of one or more cranial sutures that leads to the distortion of the skull shape. It is a frequent development anomaly, with an estimated incidence of 1 in 2,100 to 2,500 children. Syndromic craniosynostosis accounts for 30% of the cases which are classified in more than 150 syndromes [ 1,2]. Within these, there is a group of acrocephalosyndactyly syndromes (ACS) which include: Saethre-Chotzen, Crouzon, Baller-Gerold, Apert, and Pfeiffer. These syndromes present with cranial and limb abnormalities. Since these syndromes share some clinical features, establishing a specific clinical diagnosis can be challenging. For this reason, we present the case of a classical Saethre-Chotzen syndrome (SCS) which diagnosis was made clinically. We demonstrate how the diagnosis was achieved through the comparison of the patient’s characteristics with the distinguishing features of each ACS syndrome.
Case Report
A 29-year-old male was referred to the genetics department for the evaluation of cranial and limb malformations. He was product of the first pregnancy, born at 37 weeks, following an uneventful pregnancy of non-consanguineous parents. His father had similar phenotypic features, nevertheless, no diagnostic evaluation was ever performed. The patient has no perinatal pathological history and had a normal psychomotor development. He has had three hand surgeries with the purpose of establishing a phalangeal pincer grasp due to partial cutaneous syndactyly of the 2nd and 3rd digit of the left hand and total syndactyly of the right hand.
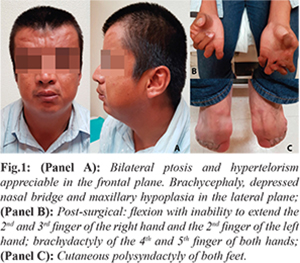
The somatometry is within normal parameters: height 176 cm (50th percentile), weight 73 kg (40th percentile) and body mass index 23.6 kg/m2. The physical examination revealed noticeable craniofacial malformations secondary to bilateral coronal craniosynostosis: brachycephaly, hypertelorism, slight bilateral ptosis, depressed nasal bridge, small pinna with prominent crus, and mild hypoplasia of the jaw without oral cavity anomalies [Fig.1, Panel A]. Both hands have post-surgical changes and significant limitation of movement [Fig.1, Panel B]. Complete simple polysyndactyly of both feet was also present [Fig.1, Panel C]. He has a preserved intellectual capacity. The patient was evaluated by cardiology, ophthalmology, and orthopedics and no other structural or functional abnormalities were found.
The findings on the skull radiographs include: brachycephaly with decreased fronto-occipital diameter, maxillary hypoplasia, and mild cortical calvarium thickening [Fig.2, Panel A]. In the cervical radiographs, no vertebral fusions are observed [Fig.2, Panel B]. The radiograph of the hands demonstrates brachydactyly of all the phalanges; there is no evidence of bone syndactyly or distal radial hypoplasia [Fig. 2, Panel C]. The radiograph of the feet reveals duplicated proximal phalanges of the 2nd toe and bifid hallux of both feet; there is also complete soft tissue syndactyly but no evidence of bone syndactyly [Fig.2, Panel D].
Discussion
Saethre-Chotzen syndrome (SCS) is an autosomal dominant acrocephalosyndactyly (ACS) syndrome, with an estimated incidence of 1 in 25,000 to 50,000 live births [ 1]. Mutations of the TWIST1 gene in chromosome 7p21 has been detected in 60-80% of SCS patients [ 1,2]. There have been several mutations described including: gene deletions, intragenic nonsense and frameshift mutations. However, a correlation between the genotype and phenotype has not been described, except for large deletions which present a higher risk of learning disabilities [ 3]. SCS presents with craniosynostosis, usually of the coronal sutures, which causes craniofacial dimorphism that include: brachycephaly, hypertelorism, low frontal hairline, and small pinna with prominent crus. It is also frequently associated with limb abnormalities such as: brachydactyly, syndactyly of the second and third digit, bifid hallux, and short stature. Most patients develop normal intellectual skills [ 2].
Despite the phenotypic variability of the SCS, it is possible to establish a diagnosis based on the clinical findings. This patient presents the distinctive features of this syndrome; the most evident is the brachycephaly suggestive of bilateral coronal craniosynostosis. The alteration in shape of the cranial vault varies with the fused sutures, so that compensatory growth occurs in dimensions not restricted by sutures [ 4]. The patient has small ears with prominent crus, bilateral ptosis and low frontal hair implantation, which are also specific malformations of the SCS. Limb abnormalities are not found in all cases but when present, like the syndactyly and bifid hallux seen in this patient, they are highly suggestive of SCS [ 5]. Bone malformations, such as maxillary hypoplasia, hypertelorism, and fusion of vertebral bodies are also features that distinguish this syndrome. In this patient, we can identify the first two traits; however, the fusion of vertebral bodies is not present, this finding is usually described in more severe phenotypes [ 6,7].
Since many of the phenotypic characteristics may overlap, it is important that when assessing a patient with an ACS syndrome, all the differential diagnoses are considered. Despite their similarities, there are specific characteristics of each syndrome that help us to differentiate between them. Crouzon syndrome is characterized by bicoronal craniosynostosis and hypoplasia of the facial bones which present with more severe facial deformity and proptosis, due to the recoil of the maxilla and the forehead [ 8]. Baller-Gerold syndrome also presents with bilateral coronal craniosynostosis and other characteristics such as proptosis, oligodactyly and aplasia/hypoplasia of the radius, which were not present in this patient [ 7]. Apert syndrome (AS), like Crouzon, presents with a more severe facial hypoplasia, hooked nose and proptosis. In addition, the syndactyly seen in AS patients is usually cutaneous and osseous. Pfeiffer syndrome distinguishing feature is the widening and medial deviation of the thumbs and first toes, which in not seen in this patient [ 9]. Finally, another important differential diagnosis of SCS is Muenke syndrome (MS), a coronal craniosynostosis with brachydactyly which distinct feature is carpal and/or tarsal bone coalition [ 1]. MS facial deformities are similar to those seen in SCS, however, patients with MS do not present low hairline, ptosis, malformations of the ears or cutaneous syndactyly. In addition, the risk of intellectual disability is greater in MS than in SCS [ 10].
Conclusion
The differential diagnosis in ACS syndromes is generally based on the presence or absence of distinct limb and facial features due to wide phenotypic variations in patients with identical mutations. The clinical diagnosis is of great importance, particularly in medical practice scenarios where resources are limited. In those cases, in which there is no access to molecular testing, the physical examination and radiographic findings lead to the diagnosis. However, in very mild or atypical cases where phenotypic variability does not lead to a specific diagnosis, molecular analysis should be used to confirm the clinical suspicion [ 11].
Contributors: APSB, IPDR: manuscript writing, case management and intellectual input; PRM, JFSG: manuscript editing, literature search, photographs and case management. APSB will act as guarantor. All authors approved the final version of the manuscript. Funding: None; Competing interests: None stated.
References - Wang JC, Nagy L, Demke JC. Syndromic Craniosynostosis. Facial Plast Surg Clin North Am. 2016;24:531-543.
- Ko JM. Genetic Syndromes Associated with Craniosynostosis. J Korean Neurosurg Soc. 2016;59:187-191.
- Spaggiari E, Aboura A, Sinico M, Mabboux P, Dupont C, Delezoide A-L, Guimiot F. Prenatal diagnosis of a 7p15-p21 deletion encompassing the TWIST1 gene involved in Saethre–Chotzen syndrome. Eur J Med Genet. 2012;55:498-501.
- Vlad Ciurea A, Toader C. Genetics of craniosynostosis: review of the literature. J Med Life. 2009;2:5-17.
- Trusen A, Beissert M, Collmann H, Darge K. The pattern of skeletal anomalies in the cervical spine, hands and feet in patients with Saethre–Chotzen syndrome and Muenke-type mutation. Pediatr Radiol. 2003;33:168-172.
- Gripp KW, Stolle CA, Celle L, McDonald-McGinn DM, Whitaker LA, Zackai EH. TWIST gene mutation in a patient with radial aplasia and craniosynostosis: further evidence for heterogeneity of Baller-Gerold syndrome. Am J Med Genet. 1999;82:170-176.
- Seto ML, Lee SJ, Sze RW, Cunningham ML. Another TWIST on Baller-Gerold syndrome. Am J Med Genet. 2001;104:323-330.
- Chico F. Craneoestenosis. II. Análisis de las craneoestenosis sindromáticas y diferentes tipos de tratamiento. Bol Med Hosp Infant Mex. 2011;68:409-418.
- Reardon W, Winter RM. Syndrome of the month Saethre-Chotzen syndrome. Jf Med Genet. 1994;31:393-396.
- Gallagher ER, Ratisoontorn C, Cunningham ML. Saethre-Chotzen Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al. Bird TD, Fong CT, Mefford HC, Smith RJH SK, (ed). Gene Reviews(®). Seattle, WA: University of Washington, Seattle;1993-2016.
- Wilkie AOM, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, et al. Prevalence and Complications of Single-Gene and Chromosomal Disorders in Craniosynostosis. Pediatrics. 2010;126:391-400.
|
|
|
|
|
|
|
Search Google Scholar for
|
|
|
Article Statistics |
|
Soto Brambila AP, Moragrega PR, Dávalos Rodríguez IP, Santoscoy Gutiérrez JFAcrocephalosyndactyly Syndrome: Challenging Diagnosis.JCR 2017;7:204-207 |
|
Soto Brambila AP, Moragrega PR, Dávalos Rodríguez IP, Santoscoy Gutiérrez JFAcrocephalosyndactyly Syndrome: Challenging Diagnosis.JCR [serial online] 2017[cited 2026 Jun 4];7:204-207. Available from: https://www.casereports.in/articles/7/2/Acrocephalosyndactyly-Syndrome.html |

|
|
|
|
|