|
|
|
|
|
Unusual Presentation of Griscelli Syndrome: Neurological Involvement without Hematologic Manifestations
|
|
|
|
Shalini Akunuri, Akanksha Jain, Varun Narwal, Shahzad Alam Department of Pediatric Intensive care, Narayana Health, Bangalore, India. |
|
|
|
|
|
Corresponding Author:
|
|
Dr. Shalini Akunuri Email: akunurishalini@gmail.com |
|
|
|
|
|
|
|
|
Received:
24-JAN-2017 |
Accepted:
21-MAR-2017 |
Published Online:
15-APR-2017 |
|
|
|
|
|
|
|
Abstract
|
|
|
|
Background: Griscelli syndrome type-2 (GS2) is characterized by cutaneous hypopigmentation, immunodeficiency, and hemo-phagocytic lympho-histiocytosis. Primary neurological presentation is rare. Case Report: We hereby report 13 year old girl who presented with cutaneous hypopigmentation, clinical and radiological evidence of neurologic involvement unaccompanied by other features of hemo-phagocytosis. Electron microscopy of hair shaft and skin biopsy established the diagnosis of Griscelli syndrome. Conclusion: Neurologic manifestations in Griscelli syndrome may occur in isolation without the more common hemophagoctic-lymphohistiocytosis. |
|
|
|
|
|
Keywords :
|
Biopsy, Hair, Hypopigmentation, Immunologic Deficiency Syndromes, Lymphohistiocytosis.
|
|
|
|
|
|
|
|
|
|
|
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffa4ca17000000de02000001001e00 6go6ckt5b5idvals|743 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Griscelli syndrome is a rare autosomal recessive disorder resulting in pigmentary dilution of the skin and hair, presence of large clumps of pigment in hair shafts and an accumulation of melanosomes in melanocytes [ 1]. Griscelli syndrome type 2 (GS2) is characterized by partial albinism, immunodeficiency, organomegaly and accelerated phases [ 2]. During the accelerated phases, hemo-phagocytosis and pancytopenia occur which may be accompanied by neurological deterioration [ 2, 3]. Isolated neurological deterioration without hematological involvement has been rarely described [ 4- 6]. We describe a girl who presented with isolated neurological manifestations. The other unusual finding in this child was the presence of pulmonary nodules which has not been described earlier, making the case a diagnostic challenge.
Case Report
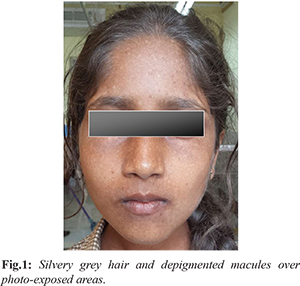
A 13 year old girl presented with progressive ataxia, intentional tremors and slurring of speech for two months. She was noted to have silvery grey hairs, eyelashes and eyebrows since birth. Physical examination revealed above findings with de-pigmented macules and freckles over photo-exposed areas (face, arms, legs and neck) [Fig.1]. She had hyper-reflexia, ataxia, dysdiadochokinesis, past pointing and intentional tremors which were suggestive of cerebellar involvement. However there were no neurological deficits and rest of the systemic examination was unremarkable. She was third child of consanguineous parents (1st cousins), with the other two siblings being normal. The child’s great-grandmother also had silvery grey hair since birth, though she never suffered any systemic illness.
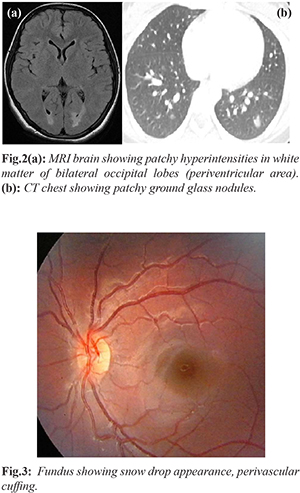
Laboratory data revealed normal complete blood counts, peripheral smear, liver and renal function test and mildly elevated ESR. Sepsis workup and CSF study including cytospin for malignant cells were negative. LDH was mildly elevated and serum ferritin, fibrinogen levels, triglycerides were normal ruling out possibility of hemo-phagocytosis [Table 1]. MRI brain was done which showed patchy hyper-intensities in white matter of bilateral cerebellar hemispheres and occipital lobes (periventricular area) [Fig.2a]. DNA PCR for acid fast bacillus was negative. Ophthalmic examination revealed snow drop appearance, perivascular cuffing [Fig.3] and few pigmented iris spots.
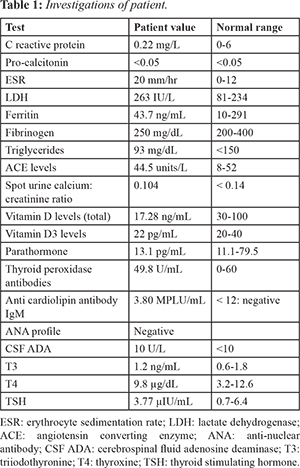
In view of wide spread skin lesions, neurologic manifestations, perivascular cuffing on eye examination and pulmonary nodules, childhood onset neuro-sarcoidosis was also considered, however evaluation for the same-total vitamin D and D3 levels, parathormone, lymphnode biopsy, spot urine calcium creatinine ratio were within normal limits. Mantoux, anti-nuclear antibody (ANA), anti-mitochondrial antibody (AMA) M2 IgG, anti-cardiolipin antibody, and angiotensin converting enzyme (ACE) levels were normal [Table 1]. Chest x-ray was normal. However, CT chest showed multiple patchy areas of ground glass density nodules in both lungs [Fig.2b]. Ultrasonography abdomen and pelvis was normal. Lymph node biopsy done to demonstrate presence of any sarcoid granules was negative.
Characteristic silvery grey hairs and skin lesions, history of consanguinity and family history of similar skin lesions led us to consider a possibility of Griscelli syndrome. Microscopic hair shaft examination was done, which revealed large unevenly distributed melanin aggregates along the length of the hair shaft mainly near the medulla, diagnostic of Griscelli’s syndrome. Skin biopsy [Fig.4] showed clumps of enlarged melanocytes in the basal layer. A diagnosis of neuro-accelerated phase of Griscelli-2 was considered.
Discussion
Griscelli syndrome is characterized by partial pigmentary dilution with silvery grey hairs, cellular immune deficiency, neurologic abnormalities and uncontrolled T-lymphocyte and macrophage activation. GS is classified into 3 types based on genetic and molecular features. GS1 is related to MYO5A gene, and is described as silvery gray hair, severe neurologic symptoms at birth without any signs of accelerated disease. GS3 manifestations are restricted to skin and hair and results from mutations in the gene that encodes melanophilin (Mlph) [ 7]. GS2 caused by mutations in RAB27A is characterized by hypopigmentation associated with an immune defect, leading to episodes of life-threatening uncontrolled T-lymphocyte and macrophage activation that are the hallmark of hemophagocytic lymphohistiocytosis (HLH) [ 8]. During accelerated phase, various organs are infilterated, including central nervous system. Sometimes neurologic manifestations may be the first sign of an accelerated phase. Neurologic involvement with raised intracranial pressure, cerebellar signs, encephalopathy, hemiparesis, peripheral facial palsy, spasticity, hypotonia, seizures, psychomotor retardation and progressive neurologic deterioration is known [ 3]. Neurologic involvement is secondary to diffuse lympho-histiocytic organ infiltration or to undocumented viral brain infection [ 9].
The patient described by us presented in an unusual manner with a purely neurological picture without any hematological abnormalities or hepato-splenomegaly. Such cases of GS2 with isolated neurological involvement in the absence of hematological manifestations of HLH have rarely been reported [ 4- 6]. The other unusual finding in this child was the presence of pulmonary nodules, which has not been described with Griscelli syndrome before; rather, this finding shifted the focus to other conditions like neurosarcoidosis making the diagnosis a clinical challenge.
The main differential diagnoses for GS are Chediak-Higashi syndrome (CHS) and Elejalde syndrome (ES). Both disorders can present with the characteristic silvery grey hair, eyebrows and eyelashes. The hallmark of CHS is the presence of giant intra-cytoplamic granules in virtually all granulated cells, which is never observed in GS10. In CHS, the hair shaft also contains a typical pattern of uneven accumulation of large pigment granules but in GS the clusters of melanin pigment on the hair shaft are six times larger than in CHS [ 10]. Elejalde syndrome, like GS has presence of spotty hair pigmentation, but incomplete melanization of melanosomes in skin, and no immunodeficiency [ 12].
Neuroimaging findings consist of cerebellar hypodense areas, ventricular dilation, hyperdense areas compatible with inflammatory changes, white matter changes and periventricular calcifications [ 8]. Cortical-subcortical atrophy and hypointensity in bilateral temporal white-matter in MRI was documented by Alva-Moncayo et al. [ 13]. Similar lesions of patchy hyper-intensities in white matter of bilateral cerebellar hemispheres and occipital lobes were noted in our case also. Although our patient did not present with HLH, however it could manifest in later life. In patients with GS1, there is no definitive cure, GS2 has a grave prognosis and bone marrow transplantation is the only curative treatment, while GS3 does not pose a threat to those affected and needs no active intervention.
Conclusion
Neurologic manifestations as a part of accelerated phase in Griscelli Syndrome may occur in isolation without the more common hemophagocytic lymphohistiocytosis. However close monitoring for the development of hematologic manifestations is essential.
Contributors: SA: Report conceptualization, manuscript drafting, literature search; AJ: literature search and inputs into the manuscript; SA,VN: Manuscript reviewing and editing. SA and VN were involved in clinical care of the patient. SA will act as guarantor. All authors approved the final version of the manuscript. Funding: None; Competing interests: None stated.
References - Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscelli syndrome: Report of a case and review of the literature. J Am Acad Dermatol. 1998; 38:295-300.
- Meschede IP, Santos TO, Izidoro-Toledo TC, Gurgel-Gianetti J, Espreafico EM. Griscelli syndrome-type 2 in twin siblings: case report and update on RAB27A human mutations and gene structure. Braz J Med Biol Res. 2008;41:839-848.
- Hurvitz H, Gillis R, Klaus S, Klar A, Gross-Kieselstein F, Okon E. A kindred with Griscelli disease: spectrum of neurological involvement. Eur J Pediatr. 1993;152:402-405.
- Aksu G, Kutukculer N, Genel F, Vergin C, Omowaire B. Griscelli syndrome without hemophagocytosis in an eleven-year-old girl: expanding the phenotypic spectrum of RAB27A mutations in humans. Am J Med Genet A. 2003;116A:329-333.
- Masri A, Bakri FG, Al-Hussaini M, Al-Hadidy A, Hirzallah R, de Saint Basile G, et al. Griscelli syndrome type 2: a rare and lethal disorder. J Child Neurol. 2008;23:964-967.
- Rajadhyax M, Neti G, Crow Y, Tyagi A. Neurological presentation of Griscelli syndrome: obstructive hydrocephalus without haematological abnormalities or organomegaly. Brain Dev. 2007;29:247-250.
- Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5AF exon deletion (GS1) J Clin Invest. 2003;112:450-456.
- Blanche S, Caniglia M, Girault D, Landman J, Griscelli C, Fischer A, et al. Treatment of haemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation: a single centre study of 22 cases. Blood. 1991;78:51-54
- Klein C, Phillippe N, Le Deist F, Fraitag S, Prost C, Durandy A, et al. Partial albinism with immunodeficiency (Griscelli syndrome). J Pediatr. 1994;125:886-895.
- Fukai K, Ishii M, Kadoya A, Chanoki M, Hamada T. Chediak-Higashi syndrome: report of a case and review of the Japanese literature. J Dermatol. 1993:20:231-237
- Sheela SR, Latha M, Susy JI. Griscelli Syndrome: Rab 27a mutation. Indian Pediatrics 2004;41:944-947.
- Elejalde BR, Holguin J, Valencia A, Gilbert EF, Molina J, Martin G, et al. Mutations affecting pigmentation in man: I, Neuroectodermal melanolysosomal disease. Am J Med Genet. 1979;3:65-80.
- Alva-Moncayo E, Castro-Tarin M, Perez-Casillas R. Síndrome de Griscelli em México. descripción de um caso com manifestaciones neurológicas. Rev Neurol. 2003;36:347-350.
|
|
|
|
|
|
|
Search Google Scholar for
|
|
|
Article Statistics |
|
Akunuri S, Jain A, Narwal V, Alam SUnusual Presentation of Griscelli Syndrome: Neurological Involvement without Hematologic Manifestations.JCR 2017;7:139-142 |
|
Akunuri S, Jain A, Narwal V, Alam SUnusual Presentation of Griscelli Syndrome: Neurological Involvement without Hematologic Manifestations.JCR [serial online] 2017[cited 2026 Jun 1];7:139-142. Available from: http://www.casereports.in/articles/7/2/Unusual-Presentation-of-Griscelli-Syndrome.html |

|
|
|
|
|