6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffa49a1f0000002b07000001000200
6go6ckt5b5idvals|826
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC), also known as arrhythmogenic right ventricular dysplasia, is a heritable heart-muscle disorder that predominantly affects the right ventricle. ARVC is one of the leading causes of arrhythmic cardiac arrest in young people and athletes. First it was described by Marcus and colleagues in 1982, describing 24 affected patients [2]. The disease was initially designated as a dysplasia because it was thought to be a congenital defect in the development of the right ventricular myocardium. The subsequent discovery that the disease is caused by a genetic defect in the cardiac desmosomes has led to its recognition as a cardiomyopathy and its inclusion in the classification of cardiomyopathies by the American Heart Association [3]. We present a clinical case of arrhythmogenic right ventricular cardiomyopathy associated with partial hypopituitarism from previous intracranial surgery.
Case Report
A 25-year-old female presented with dyspnea for last one year. She was diagnosed as systemic lupus erythematosus by a hospital in our city one year ago. Her symptoms included fatigue, weakness, inability to lose weight (or weight gain), puffiness, and constipation. She did not attain menarche. Physical examination showed periorbital puffiness, brittle hair and eyebrow loss. She had a past history of brain tumor 17 years ago at the age of 8 years. Her treatment for brain tumor was surgery, chemotherapy and radiation therapy. Subsequently she stopped taking medicines and treatment plan as per her family decision.
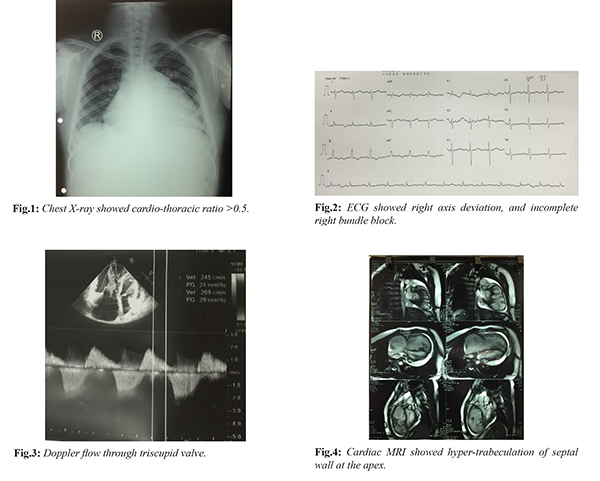
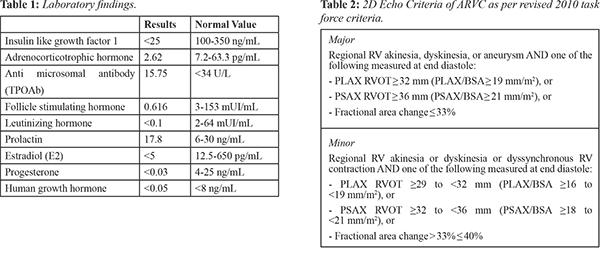
Chest X-ray demonstrated increased heart-thorax index [Fig.1]. ECG showed right axis deviation, and incomplete right bundle block [Fig.2]. This prompted further endocrine workup that revealed the presence of hypoadrenalism, hypogonadism and growth hormone deficiency. Partial hypopituitarism from previous intracranial surgery was diagnosed [Table 1]. Transthoracic echocardiogram showed normal left ventricular systolic function with ejection fraction of 53%, normal size and wall thickness of the left ventricle, slightly dilated left atrium, enlargement of the right ventricle 50 mm and right ventricular outflow tract 47 mm with reduced contractile function, tricuspid annular plane systolic excursion (TAPSE) 11 mm, apical, aneurysm-akinesia, endocardial ventricular hypertrabeculation especially at apex; mild mitral regurgitation and severe tricuspid regurgitation;and medium pericardial effusion without right ventricular depression [Fig.3]. Cardiac MRI showed normal volume and function of the left ventricle (EF 55%), right ventricle with 114 ml/m2 indexed end-diastolic volume, reduced right ventricular function (EF 29.6%), areas of dyskinesia of the free wall at the mid and apex; late gadolinium enhancementof the free wall right ventricle at the mid and apex; hypertrabeculation of septal wall at the apex; medium pericardial effusion, no cardiac tamponade [Fig.4]. ARVC was diagnosed on the basis of revised 2010 task force criteria for ARVCas mentioned in Table 2 [7].
Differential diagnosis is dilated cardiomyopathy associated with partial hypopituitarism. If hypopituitarism promptly responds to multiple hormone replacement therapy, definite diagnosis would be dilated cardiomyopathy associated with partial hypopituitarism.
Discussion
ARVC causes 11-22% of sudden cardiac death in the young athletes patient population, resulting in approximately 22% of cases in athletes in northern Italy [4] and about 17% of sudden cardiac death in young people in the United States [5]. The pathologic hallmark of disease is myocardial atrophy (myocyte loss), fibrofatty replacement, fibrosis and thinning of the wall with chamber dilation and aneurysm formation [6]. The genetics of the disease seems to support the hypothesis that it may be caused by desmosomal dysfunction (mutation of plakophilin-2, desmoglein-2, desmocollin-2, desmoplakin genes), that lead to impaired mechanical and electrical coupling between individual cells, leading to myocyte uncoupling, especially under conditions that increase myocardial strain, for example during physical effort.
Diagnosis of ARVC relies on a scoring system, formulated in 2010 by the revisited Task Force, with two major or one major and two minor criteria or four minor criteria based on the demonstration of a combination of defects in right ventricular morphology and function, characteristic depolarization/repolarization electrocardiogram abnormalities (negative T waves and/or “epsilon” waves in right precordial leads), characteristic tissue pathology, typical arrhythmias, family history, and the results of genetic testing [7]. Cardiac magnetic resonance (MRI) is considered the best imaging modality in evaluating the RV in ARVC and provides tissue characterization and identification of intra-myocardial fat and fibrosis in addition to assessment of ventricular structure and function.
Conclusion
Cardiac magnetic resonance (MRI) is considered the best imaging modality in evaluating the RV in ARVC and provides tissue characterization and identification of intramyocardial fat and fibrosis in addition to assessment of ventricular structure and function.
Contributors: HTP wrote the manuscript, did literature search, involved in patient management, and approved the final version of this article. He will act as guarantor of the study.
Funding: None; Competing interests: None stated.
References
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129-133.
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384-398.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 2006;113:1807-1816.
- Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on arrhythmogenic right ventricular dysplasia/cardiomyopathy of the working groups on myocardial and pericardial disease and arrhythmias of the European Society of cardiology and of the scientific council on cardiomyopathies of the world heart federation. Circulation. 2000;101:E101-E106.
- Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112:3823-3832.
- Thiene G, Basso C, Danieli G, Rampazzo A, Corrado D, Nava A. Arrhythmogenic right ventricular cardiomyopathy a still underrecognized clinic entity. Trends Cardiovasc Med. 1997;7:84-90.
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533-1541.