6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff24c722000000f306000001000400
6go6ckt5b5idvals|863
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Composite pheochromocytoma (CP) is a rare tumor of adrenal medulla with less than hundred cases reported in medical literature. CP has coexistent pheochromocytoma or paraganglioma with other neurogenic tumors like ganglioneuroma, ganglioneuroblastoma, neuroblastoma or peripheral nerve sheath tumors [1]. It constitutes less than three percent of all adrenal gland neoplasms [2] and sympatho-adrenal pheochromocytomas [3]. We report a rare case of nonfunctional CP-ganglioneuroma in a young girl and review relevant literature.
Case Report
A 16 year old girl presented with abdominal pain and distension of two months duration. There was no history of vomiting, diarrhea or constipation. The blood pressure was 110/70 mm of Hg. Ultrasonography and CT scan suggested right suprarenal gland mass lesion. Biochemical workup found normal ranges of serum electrolytes and cortisol levels. She had subclinical hypothyroidism with normal free thyroxin levels but elevated TSH (7.27 mg/dL). Urine metanephrine levels were 176.83 µg/24 hours. Right adrenalectomy was done; postoperative period was uneventful.
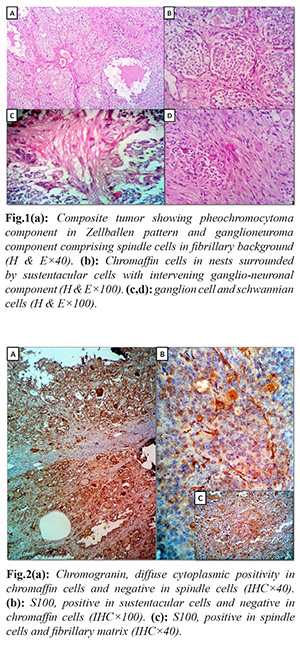
Adrenalectomy specimen measured 11×8×5 cm and weighed 346 grams. External surface was smooth and grey brown. Cut surface showed an encapsulated solid grey brown tumor with focal grey white and dark brown areas. Microscopy of Hematoxylin and eosin sections revealed an encapsulated tumor comprising two distinct components. The major component was composed of nests of polygonal chromaffin cells arranged in typical Zellballen pattern, admixed with ganglion cells and surrounded by spindle shaped sustentacular cells. The chromaffin cells showed amphophilic cytoplasm and moderately pleomorphic round to oval nuclei with stippled chromatin. There were foci of other component comprising spindle shaped schwannian cells in a fibrillary background. A histological diagnosis of CP was considered basing on the presence of two components throughout the tumor. On immunohistochemical (IHC) study, diffuse positivity for chromogranin was found in polygonal cells as well as ganglion cells. S100 was positive in sustentacular cells, and ganglioneuronal component. Hence the final diagnosis was concluded as CP.
Discussion
CP implies pheochromocytoma in combination with other neurogenic tumor component where as the term ‘mixed pheochromocytoma’ is used when adrenal medullary tumors are associated with adrenal cortical tumor. Ganglioneuroma is the most common neurogenic component in CP [4]. PC and ganglioneuroma originate from chromaffin cells and autonomic ganglion cells, respectively; both of these are derived from common progenitor neural crest cell [3].
CP can occur at any age with equal frequency in both genders; majority occurs between 40 to 60 years. According to world literature, the youngest and oldest ages reported are 5 years [5] and 83 years respectively [6].Youngest patient reported from India is 27 year old male [7]. Our patient is much younger, 16 year old, probably the youngest case reported from India. CP with immature components is more frequent in young individuals of 10 years age [2].
CPs reported in literature are mostly unilateral; they can be bilateral when associated with neurofibromatosis type 1. CP can rarely be extra-adrenal; so far twelve cases are reported in literature [4]. Clinical symptoms depend on whether it is functional or non-functional. Functional evidence is seen in three fourths of reported CPs [3]. Catecholeamine secreting tumors present with symptoms of palpitations, headache, excessive perspiration and elevated urinary VMA levels [3].Frequency of hypertension in CP ranges from 47.9 to 72.4% [3]. The present case is a non-functional tumor with normal urinary metanephrines. CPs with normal VMA levels can show functional evidence when catecholeamines secreted by PC are metabolized and modified by ganglioneuromatous component [7]. VIPs secreting CPs present with watery diarrhea, hypokalemia and achlorhydria.
Grossly, CP also is encapsulated; PC appears grey brown and darkens on exposure to air. Careful examination of cut surface can give clues to the presence of other components. The ganglioneuromatous component appears as patchy firm grey white areas while ganglioneuroblastoma areas are usually cystic, necrotic and haemorrhagic [1]. Hence, it is important to take thin slices of tumor and study all the representative areas to identify additional components.
Microscopically typical PC is admixed with other neurogenic component. Ganglioneuroma component is composed of mature ganglion cells and schwannian stroma where as ganglioneuroblastoma is composed of mature ganglion cells and immature neuroblasts. However, mere presence of ganglion cells will not qualify a pheochromocytoma into a CP as scattered ganglion cells can be seen in pure pheochromocytoma [8]. The diagnosis of CP should be reserved when both histo-architectural and cell populations of an additional tumor type are present admixed with pheochromocytoma.
Immunohistochemically, pheochromocy- toma is positive for chromogranin, synaptophysin, vimentin, neurofilament J, betatubulin, micro-tubule associated protein, GFAP, and peanut agglutin. Neuroblastoma component is also positive for all the above markers except vimentin, and GFAP, but stains less intensely as they have less secretory granules [9]. This differential staining of chromogranin and synaptophysin in chromaffin cells and neuroblastoma cells is useful in distinguishing neurogenic component in CP. Both PC and neuroblastoma are negative for S-100; sustentacular cells and schwann cells are positive for S100 [1].
Prognostic factors of pheochromocytoma include Ki 67 immunoreactivity, histological pattern, cellularity, coagulative necrosis, vascular/ capsular invasion and type of catecholeamine production. Ki 67 index of >5% is indicative of malignant behavior [2]. Prognosis of CP with GN and without immature components is similar to pheochromocytoma [1]. Shawa et al. have reported 8% rate of malignancy in CP with GN similar to typical pheochromocytoma [6]. Presence of distant metastasis is the only criterion for malignancy in CP which is usually derived from immature component [1]. The present case showed no evidence of vascular invasion or metastatic spread. Fujiwara et al. reported no recurrence in completely excised cases after five year follow up [10]. However, Nigawara et al. reported one patient who developed local recurrence after 15 years [11]. Long term follow up is required in CP.
Conclusion
This paper highlights the clinico-pathological aspects of CP with an overview of intricate diagnostic criteria and prognostic factors of composite tumors of adrenal medulla.
Contributors: KK: manuscript writing, patient management; CPD: manuscript editing, patient management; MV: critical inputs into the manuscript. CPD will act as guarantor. All authors approved the final version of this manuscript.
Funding: None; Competing interests: None stated.
References
- Tischler AS, Kimura N, Llyod RV, Komminoth P. Composite pheochromocytoma or paraganglioma. In: DeLellis RA, Lloyd RV, Heitz PU, Eng C. (Eds.): World Health Organization classification of tumours. Pathology and Genetics of Tumours of Endocrine Organs. IARC Press: Lyon 2004.
- Shida Y, Igawa T, Abe K, HakariyaT, Takehara K, Onita T, et al. Composite pheochromocytoma of the adrenal gland: a case series. BMC Res Notes. 2015;8:257.
- Rao RN, Singla N, Yadav K. Composite pheochromocytoma-ganglioneuroma of the adrenal gland: A case report with immunohistochemical study. Urology Annals. 2013;5:115-118.
- Hu J, Wu J, Cai L, Jiang L, Lang Z, Qu G, et al. Retroperitoneal composite pheochromocytoma-ganglioneuroma: a case report and review of literature. DiagnPathol. 2013;8:63.
- Tatekawa Y, Muraji T, Nishijima E, Yoshida M, Tsugawa C. Composite pheochromocytoma associated with adrenal neuroblastoma in an infant: a case report. J Pediatr Surg. 2006;41:443-445.
- Shawa H, Elsayes KM, Javadi S, Sircar K, Jimenez C, Habra MA. Clinical and radiologic features of pheochromocytoma/ ganglioneuroma composite tumours: a case series with comparative analysis. Endocr Pract. 2014;20:864-869.
- Menon S, Mahajan P, Desai SB. Composite adrenal medullary tumour: A rare cause of hypertension in a young male. Urology Annals. 2011;3:36-38.
- Linnoila RI, Keiser HR, Sternberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathological study of 120 cases including unusual histological features. Human Pathol. 1990;21:1168-1180.
- Franquemont DW, Mills SE, Lack EE. Immunohistochemical detection of neuroblastomatous foci in composite adrenal pheochromocytoma-neuroblastoma. Am J Clin Pathol. 1994;102:163-170.
- Fujiwara T, Kawamura M, Sasou S, Hiramori K. Results of surgery for a compound adrenal tumour consisting of pheochromocytoma & ganglioneuroblastoma in an adult: 5 years follow-up. Intern Med. 2000;39:58-62.
- Nigawara K, Suzuki T, Tazawa H, Funyu T, Yagihashi S, Yamaya K, et al. A case of recurrent malignant pheochromocytoma complicated by watery diarrhea, hypokalemia, achlorhydria syndrome. J Clin Endocrinol Metab. 1987;65:1053-1056.