6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffa47c24000000ad03000001002200
6go6ckt5b5idvals|885
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Thrombotic microangiopathies (TMA) are a group of microvascular occlusive disorders characterized by thrombocytopenia, hemolytic anemia and organ injury of variable cause that requires specific treatment [
1,
2]. TMA syndromes can be primary or secondary with diverse clinical presentation. Although TMA has varied etiology and clinical presentation they are united by common hematologic and pathological findings in the various organs. In this article we summarize three cases of histologically proven TMA of varying etiologies.
Case Report
Case-1
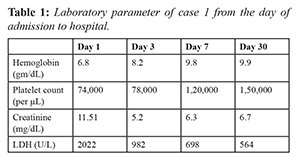
A 25 years old male presented with the history of easy fatigability and reduced appetite of two weeks duration. On evaluation he was found to have pallor, hypertension with grade II hypertensive retinopathy and bilateral pedal edema. Laboratory evaluation showed thrombocytopenia, azotemia (creatinine of 11.51 mg/dL) and features of hemolytic anemia characterized by indirect hyper-bilirubenemia and elevated lactate dehydrogenase (LDH) levels. Evaluation of bicytopenia with peripheral smear showed schistocytes and fragmented RBCs. Renal evaluation showed normal sized kidney with bland urinary sediment for which he underwent renal biopsy which showed features suggestive of thrombotic microangiopathy. He underwent further evaluation for etiology of TMA such as viral markers, autoimmune workup which were negative. His complement levels C3 was low and C4 was normal and anti-complement factor H (CFH) antibody testing was negative. He was diagnosed to have complement mediated hemolytic uremic syndrome (HUS) and was managed with plasma-exchange and hemodialysis. After starting plasmapheresis, his platelet count and LDH levels improved however he remained dialysis dependent. He discontinued plasma exchange after five sessions and was not affordable for eculizumab (monoclonal antibody against complement C5) which is the drug of choice for complement mediated TMA. At one year of follow up, he is on maintenance hemodialysis with good functional status.
Case 2
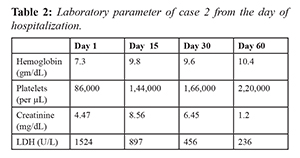
A 35 years old male presented with easy fatigability, headache, and breathlessness of one week duration. On evaluation he was found to have uncontrolled blood pressure (BP: 230/130 mm Hg) with grade IV hypertensive retinopathy, bilateral pedal edema, bilateral basal crepitations. Laboratory evaluation showed anemia, azotemia (creatinine 4.57 mg/dL) and evidence of hemolytic anemia. Evaluation of renal failure showed normal sized kidney with sub-nephrotic proteinuria. He was managed with anti-hypertensives and hemodilaysis. After adequate control of blood pressure he underwent renal biopsy which showed features suggestive of TMA. He was diagnosed to have secondary TMA probably precipitated by uncontrolled hypertension. He was continued on maintenance hemodialysis through tunnelled hemodialysis catheter and blood pressure was controlled with anti-hypertensive medication. On second month follow up his renal function recovered and he discontinued hemodialysis. He underwent detailed evaluation to rule out other possible causes of TMA including complement level and autoimmune work up which were negative. Evaluation for secondary hypertension was done including renal Doppler, dexamethasone suppression test, metanephrine levels, thyroid stimulating hormone, which was within normal range. At one year follow up he is on single anti-hypertensive medication and his renal function is normal.
Case 3
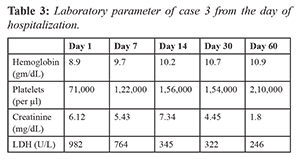
A 74 year old male who is a known case of type 2 diabetes mellitus, hypertension since five years presented with acute febrile illness, bloody diarrhea and easy fatigability of two weeks duration. General physical examination showed pallor, normal blood pressure and bilateral pedal edema. On evaluation, he was found to have anemia, leucocytosis, thrombocytopenia, and renal failure. Evaluation of renal failure showed normal kidney size with bland urinary sediment. In view of worsening of renal failure and hyper-kalemia, he was started on hemodialysis. Blood culture and urine culture was sterile. Test for tropical infection, serological and immunological test were negative. In view of hemolytic anaemia, thrombocytopenia and renal failure, a provisional diagnosis of hemolytic uremic syndrome was made. ADAMTS13 (A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity was within normal limits. His stool sample was positive for shiga toxin. He was diagnosed to have shiga toxin producing Escherichia coli hemolytic uremic syndrome (STEC-HUS). He was managed with hemodialysis and supportive measures. In view of persistent renal failure he underwent renal biopsy which showed features suggestive of thrombotic microangiopathy. On follow up his renal function partially recovered and he discontinued dialysis after two months. Evaluation for autoimmune disease and complement abnormalities were performed which were normal.
Discussion
We can observe from the above cases that, the clinical presentation of TMA can be of diverse nature such as renal failure, malignant hypertension or bloody diarrhea. All of above cases had a common finding of microangiopathic hemolytic anemia (MAHA), thrombocytopenia and acute kidney injury (AKI). TMA has diverse clinical features which are unified by histological findings, although histological feature are not necessary for diagnosis. Historically TMA was classifies as thrombotic thrombocytopenic purpura (TTP) with predominant neurological involvement and HUS with predominant kidney involvement. The understanding of molecular mechanism lead to the specific terminology of TTP for ADAMTS13 deficiency and STEC-HUS (Shiga toxin producing Escherichia coli associate HUS) for shiga toxin mediated HUS [
3]. Non STEC-HUS was called as atypical HUS, however recent understanding of pathogenesis of atypical HUS has resulted in renaming a subset as complement mediated HUS in patient with alternative complement pathway defect [
4]. Current classification describes as primary TMA either inherited (complement mutations, ADAMTS13 deficiency) or acquired (autoantibody to complement factor H or ADAMTS13 autoantibody), secondary TMA and infection associated TMA. Secondary TMA include those associated with autoimmune diseases (SLE, anti-phopholipid antibody syndrome, vasculitis), bone marrow transplantation, malignancy, malignant hypertension and drugs [
5].
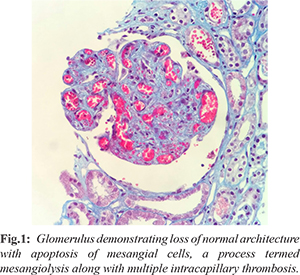
Histological features of thrombotic microangiopathy
[Fig.1] in the kidney include mesangiolysis, crumpling of basement membrane, endothelial cell swelling with or without platelet-fibrin thrombi indicating endothelial cell injury [
6]. In case one, patient was diagnosed to have complement mediated TMA as patient presented with MAHA, renal failure and low complement (C3). Complement activation and dysregulation is central to the pathophysiology of complement mediated TMA (also called atypical hemolytic uremic syndrome). In around 5-10% of patients with aHUS, auto-antibodies to factor H (CFH) have also been implicated as triggering factor. The prevalence of CFH antibody is much higher in Indian population compared to Western population. CFH antibody positive patients can be treated with immunosuppression in addition to plasmapheresis. Further evaluation requires genetic studies to look for mutation in complement factor H and I (CFH and CFI)) which was not done due to financial constraints. These complement mutation results in continuous activation of alternate complement pathway which results in lysis of target cells such as red blood cells, platelets and endothelium with organ injury. Treatment of complement mediated TMA include eculizumab (complement C5 inhibitor) which is very expensive.
In case two, patient presented with malignant hypertension with heart failure, papilledema and renal failure. Malignant hypertension can be associated with MAHA, thrombocytopenia and histological evidence of TMA on biopsy. As our patient recovered his renal function after control of blood pressure, he was diagnosed as secondary TMA precipitated by severe hypertension. Most of the secondary TMA patients usually have some complement mutation and hypertension acts as a precipitating factor. Although our patients did not undergo complement mutation testing, his serum complement levels are normal. He underwent evaluation for secondary hypertension which was found to be negative. Most likely cause of hypertension would be essential hypertension as it’s the most common cause of malignant hypertension.
In case three, we had elderly male presenting with diarrhea and features of TMA. He was positive for shiga toxin and was diagnosed to have STEC-HUS. About 5% of STEC-TMA may not have diarrhea and about 30% of complement HUS can have concurrent diarrhea [
7]. STEC-HUS is common than complement mediated HUS and usually seen in children. STEC HUS can affect adults and usually has high morbidity compared to children [
8]. Pathogenesis includes translocation of shiga toxin across intestinal epithelium into leucocytes in the blood. It then gets internalized to endothelial cells via Gb3 receptor and activates proteases to cause cell death [
9]. Treatment of STEC-HUS is supportive and has a good clinical outcome compared to other TMA. About 70% have complete recovery with mortality rate of 1-4% [
10].
Conclusion
TMA is a histological term encompassing microangiopathic hemolytic anemia, thrombocytopenia and organ injury of varied etiologies. TMA has diverse clinical presentation with significant morbidity and mortality. Early identification of etiology and targeted therapy such as plasmapheresis can decrease the morbidity and mortality.
Contributors: SLMR: Design of the work, collection and analysis of the case, drafting the case; PS: concept of the work, revising the work, accuracy of the work. PS will act as guarantor of study. Both authors approved the final version of this manuscript.
Funding: None; Competing interests: None stated.
References
- Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589-600.
- George JN, Nester CM. Syndrome of thrombotic microangiopathy. N Eng J Med. 2014; 37:1847-1848.
- Scully M, Cataland S, Coppo P, Dela Rubia J, Friedman KD, Hovinga K, et al. International working group for TTP: Consensus on the standardization of terminology of TTP and related thrombotic microangiopathy. J Thromb Haemost. 2017;15:312-322.
- Kerr HH, Richards A. Complement mediated injury and protection of endothelium:lessons from atypical haemolytic uremic syndrome. Immunobiology. 2002;217:195-203.
- Manenti L, Gnappi E, Vaglio A, Allegri L, Noris M, Bresin E, et al. Atypical haemolytic uremic syndrome with underlying glomerulopathies: case series and review of literature. Nephrol Dial Transplant. 2013:28:2246-2249.
- Goodship TH, Cook HT, Fakhouri F, Fervenza F, Fremeaux bacchii V, et al. Atypical haemolytic uremic syndrome and C3 glomerulopathy: Conclusions from “kidney disease and improving global outcome” (KDIGO) conference. Kidney Int. 2017;91:539-551.
- Brandt JR, Fouser LS, Watkins SL, Zelokovic I, Tarr PI, Nazar Stewart P, et al. Escherichia Coli 0157:H7 associated haemolytic uremic syndrome after ingestion of contaminated hamburger. J Paediatr. 1994;125:519-526.
- Gould LH, Demma L, Jones TF, Hurd S, Vugia DJ, Smith K, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49:1480-1485.
- Brigotti M, Caprioli A, Tozzi AE, Tazzari PL, Ricci F, Conte R, et al. Shiga toxin present in the gut and polymorphonuclear leucocyte circulating in the blood of children with haemolytic uremic syndrome. J Clin Microbiol. 2006;44:313-317.
- Spinale JM, Ruebner RL, Copelovitch L, Kaplan BS. Long term outcomes of shiga toxin haemolytic uremic syndrome. Paediatr Nephrol. 2013;28:2097-2105.