6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffe4f42a0000009206000001000800
6go6ckt5b5idvals|962
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Lysosomal storage diseases constitute a set of over 50 distinct metabolic diseases; each of the individual type is caused by a specified deficient function of lysosomal enzyme or enzyme trans-porter or abnormalities in the lysosomal biogene-sis or vesicular trafficking [
1]. Metachromatic leukodystrophy is the inherent error of metabolism caused by the functional deficiency of the lysosomal enzyme arylsulfatase A, which catalyzes the hydrolysis of sulphated glycosphingolipids, primarily sulfatides. Cells deficient in ARSA are unable to hydrolyze the sulfatides and ultimately lead to its accumulation in intra-lysosomal storage granules which upon histological staining appear as metachromatic inclusions [
1,
2]. Leukodystrophies are group of inherited metabolic disorders in which there is progressive degeneration of the myelin sheath, that act as insulators for nerve fibres [
3]. This MLD generally becomes manifested mostly between the first four years of life and sometimes at the juvenile stages or adult onset [
3].
Case Report
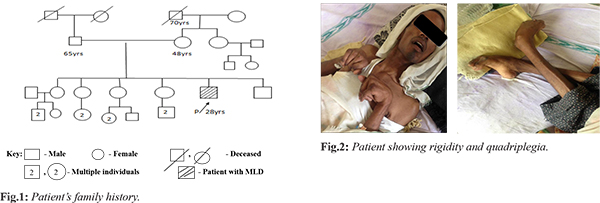
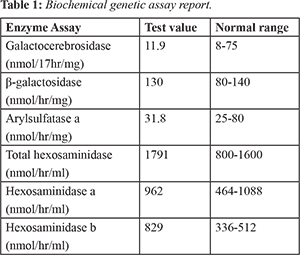
A 28 year old male patient presented with complaints of weakness of both upper and lower limbs in the form of difficulty in holding glass with left hand, dragging of left foot while walking, swaying while walking to left. This was associated with involuntary movements and intentional tremor which decreased with rest. He also had slurring of speech, blurring of vision for the past 4 months, urinary incontinence since 6 months, fever associated with chills since 5 days and behavioral abnormalities. He prefers loneliness most of the time associated with anger, irritation, suspicious looks with one episode of visual hallucination. Patient’s family history is shown in Fig.1. His MRI scan revealed symmetrical T1 hyper-intensities, T2 flair hyper-intensities involving the bilateral peri-ventricular white matter (predominantly frontal and posterior parietal), splenium and genu of corpus callosum with relative sparing of U fibers. On post-contrast images there was no significant enhancement associated with mild patchy diffusion restriction which is considered as adult onset metachromatic leukodystrophy. CT scan of brain showed hypo-density of white matter noted in central semiovale, corona radiate and adjacent to bilateral frontal horns and occipital horn of bilateral lateral ventricles. Loss of deep white matter volume with hypo-density of peri-ventricular white matter with relative sparing of subcortical U fibers was noted. Cerebellum appeared normal, mild prominent ventricular system, altogether revealing the white matter disorder. Patient was done with biochemical genetic tests 4 years back when disease was suspected and reports were normal. Biochemical genetic assay report was shown in Table 1. Presently they can’t afford these tests and were not done. Patient’s younger brother was diagnosed with adrenoleukodystrophy whereas his sisters have not shown with any of these genetic conditions. Patient was having rigidity and quadriplegia is shown in Fig.2. Current treatment plan of patient included antibiotics like ceftriaxone to prevent the hospital acquired infections, pantoprazole for gastrointestinal symptoms, optineuron and vitamin B12 for nutritional deficiencies, acetaminophen as he was suffering with fever. Patient was also given with baclofen (muscle relaxant) which was thought to reduce the slowing of axon conduction and slows the motor unit paralysis. Calcium gluconate was given as it is the necessary element for normal nerve function. Citicholine was initiated as symptomatic neuro-protective which is thought to be beneficial in slowing the neurological disorders. The neuro-protective assumption of citicholine is that it undergoes sequential hydrolysis to choline and cytidine. These metabolites enter the brain tissues and used to resynthesize the CDP-choline which exerts the neuro-protective action by intra-cellularly supporting the biosynthesis of cellular phospholipids.
Discussion
There are very few cases reported on metachromatic leukodystrophy with different findings. MLD is mainly present in three forms, they are late infantile (1-2 yrs) which is commonest, juvenile (3-10 yrs) and the adult onset (>16 yrs). It is mainly acquired by autosomal recessive inheritance pattern [
3]. The family history of patient in present case did not have any active disease and therefore most likely thought to be because of carriers.
The clinical features of the metachromatic leukodystrophy include hypotonia (low muscle tone), loss of mental abilities, blindness, speech abnormalities, dementia, mental deterioration, rigidity, impaired growth and sometimes convulsions [
3]. The present case report reveals to have adult onset MLD depending on symptoms and age of onset. The symptoms of the patient like gradual deterioration in intellectual performance, difficulty in walking, inability to speak, impaired vision, rigidity, quadriplegia and behavioral disturbances relate to that of adult onset MLD. MRI scan suggested the symmetrical hyper-intensities involving the bilateral peri-ventricular white matter predominantly at the frontal and posterior parietal regions whereas CT brain showed the hypo-densities of white matter in central semiovale, corona radiate and adjacent to bilateral frontal horns and occipital horn of bilateral ventricles concluding the white matter disorder. Both these neuroimaging studies are suggestive for the metachromatic leukodystrophy. Being an adult onset metachromatic leukodystrophy, enzyme levels may be shown normal.
There is no definite treatment for MLD, but hemopoietic stem cell transplantation (HSCT) is considered to be beneficial. Though HSCT is ineffective in arresting demyelination and nervous system involvement but it decreases the progression of the disease with stabilizing the visual and auditory functions. Other alternatives include enzyme replacement therapy (ERT) which has been successful in treating the lysosomal storage diseases. Lysosomal enzymes like ARSA are secreted and delivered to the cells via the mannose-6-phosphate pathway [
1]. Gene therapy is another approach that averts the accumulation of sulfides and put a check to motor disabilities. To decrease the subject symptoms, medications such as neuro-protective agents, muscle relaxants are added. Physiotherapy may assist in order to reduce the loss of muscle function [
6].
Conclusion
Metachromatic leukodystrophy is a dreadful disease with gradual loss of muscle movement and mental coordination causing slow progression and death within 5 years in children whereas it varies with age in adults.
Contributors: SMA, PSGR: manuscript writing, and literature review and references; RK: critical inputs into the manuscript and patient management. SMA will act as a study guarantor. All authors approved the final version of this manuscript.
Funding: None; Competing interests: None stated.
References
- Boelens JJ, Wynn RF (eds). Stem cell therapy in lysosomal storage diseases (stem cell biology and regenerative medicine). Humana Press, 2013.
- Vanier MT, Caillaud C. Disorders of sphingolipid metabolism and neuronal ceroid-lipofuscinoses. In: Saudubray JM, van den Berghe G, Walter JH. eds. Inborn metabolic diseases. Diagnosis and treatment, 5th ed. Berlin: Splinger-Verlag, 2012; pp.555-78.
- Inherited metabolic diseases of the nervous system. In: Ropper AH, Adams RD, Victor M, Brown RH, Victor M (eds). Adams and Victor's Principles of Neurology. New York: McGraw-Hill Medical Pub. Division. 2005.
- Mallikarjun K, Bhayya DP, Singh D, Shyagali TR. Metachromatic leukodystrophy: A rare case report. J Adv Dental Research. 2011;2(3).
- Metachromatic leukodystrophy. Genetics Home Reference. February 2013. Available at: http://ghr.nlm.nih.gov/condition/metachromatic-leukodystrophy. Accessed on March 19, 2019.
- Rosenber JB, Kaminsky SM, Aubourg P, Crystal RG, Sondhi D. Gene therapy for metachromatic leukodystrophy. Journal of Neuroscience Research. 2016;94:1169-1179.