|
Roberto Secchi1, Federico Meconi1, Fabiana Esposito1, Annagiulia Zizzari1, Vito Mario Rapisarda1, Ida Provenzano1, Manuela Rizzo1, Lucia Anemona2, Maria Cantonetti1 1Department of Biomedicine and Prevention, Haematology Division, 2Anatomic Pathology, Department of Biomedicine and Prevention, University of Tor Vergata, Viale Oxford 81, Rome, Italy.
Corresponding Author:
Dr Roberto Secchi Email: robsecchi922@gmail.com
Abstract
Background: Lymphomas with Myc and Bcl2 and/or Bcl6 rearrangements, commonly defined as Double hit Lymphomas (DHI) or Triple hit Lymphomas (THI) have a poor prognosis than those with diffuse large B-cell lymphoma (DLBCL) NOS. Combined Myc and Bcl-2 and Bcl-6 rearrangements are reported in rare cases of follicular lymphoma (FL) and acute lymphoblastic B-cell precursor leukemia/lymphoma (BCP-ALL). Case Report: Here in, we report a case of a fifty year-old man, who was referred to our hospital after being diagnosed with de novo DH-BCP-ALL, presenting with both diffuse cranial and spinal meningeal involvement and multiple other extra-nodal localizations (cutaneous, testicular, gastro-enteric and skeletal), in addition to nodal localizations such as para-cardiac and para-renal. After reaching Partial Response (PR) with first line treatment, the patient early relapsed and was refractory to new lines of therapy with an overall survival (OS) of 7 months. Conclusion: BCP-ALL with combination of Myc and Bcl-2 and/or Bcl-6 rearrangements named “De novo DH-BCP-ALLs” have to be classified as a separate entity although they are rare and their clinic immune-phenotypic and cytogenetic features are not well characterized.
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff648c310000007b06000001000700 6go6ckt5b5idvals|2042 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
A recent review in literature identified 6 patients in the German Multicenter Study Group for Adult LL (GMALL) registry and 11 patients published between 1983 and 2018 with De novo DH-BCP-ALLs, analyzing the common features, therapy approaches and poor outcomes [ 1]. We aim to share our personal experience and compare it to others, in order to infer what clinical and biological key point we can find in such limited cases reported.
Case Report
In April 2019, the patient presented progressive brachialgia, paresthesia, and hypotrophy of the right upper limb, associated with loss of function of the ipsilateral hand. A Magnetic Resonance (MR) of the spine showed an extradural hourglass mass, extending from C6 to D1 and displacing the spinal cord. The pathological evaluation of the lesion showed proliferation of medium lymphoid cells with "starry sky" appearance and frequent mitoses (Ki67 proliferation index 95%). Immuno-phenotype analysis showed rare Tdt positivity (10-15%), CD45/LCA+, CD79a+, PAX5+, CD20 +/-, CD34-, CD99-/+, CD10+, BCL6-, IRF4 +/-, BCL2+, IgM+, C-Myc+. Both Myc and Bcl-2 were rearranged in FISH analysis, cytogenetic analysis showed a normal karyotype. The findings were consistent with B-ALL with mature phenotype. Initial positron emission tomography (PET) computed tomography (CT) [Fig.1a,b] scan revealed marked diffuse FDG activity involving the pericardium (SUV max 10), the palatine tonsils and the tongue (SUV max 10), the stomach (SUV max 18), the right kidney (SUV max 5), both homers (SUV max 9), the left femur, the right scapula, the right testicle (SUV MAX 13) and two cutaneous regions, one on the right gluteus (SUV max 13) and the other localized above the VIII left rib (SUV max 3.7). Spinal cord was involved in both the previous analyzed mass and on a second level, from L5 to S1 (SUV max 15). At admission, laboratory studies showed normal levels of hemoglobin, platelets, white blood cells (WBC) and lactate dehydrogenase (LDH). The patient suffered from a severe hyponatremia (lowest of 110 mEq/L). Esophagogastroduodenoscopy (EGD) with biopsy [Fig.2] and testicular echography were performed, confirming the disease localizations. Bone marrow biopsy and aspirate showed no disease infiltration.
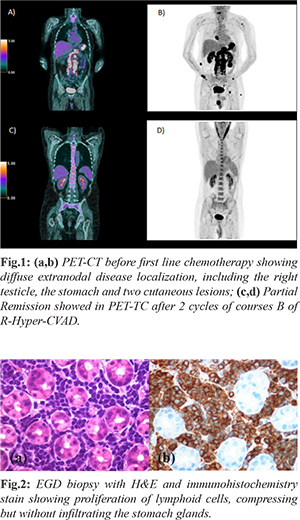
The patient underwent first line treatment with 2 cycles of course B of R-Hyper-CVAD with high dose methotrexate (3 g/mq on day 1) and cytarabine (3000 mg/mq/12 hours on day 1 and 2) and rituximab (375 mg/mq), resulting in a PR in PET-TC [Fig.1c,d]. To ensure a better response of CNS involvement, course A of Hyper-CVAD was first postponed and high dosage methotrexate was set at 3 g/mq, as in primary central nervous system (CNS) lymphoma regimen, instead of 1 g/mq (as it is in the Hyper-CVAD regimen) [ 2]. Due to the many extra-nodal sites involved, a diagnostic lumbar puncture was first performed showing meningeal disease [107 cells, 8000 events in flow cytometry (FC)] and the patient received a weekly intrathecal chemotherapy with methotrexate, cytarabine and prednisone, resulting in a progressive reduction of events in FC, without never reaching liquor sterilization. Intrathecal treatment was later interrupted due to chronic headache and liquor hypotension syndrome and never performed again. Due to the severe hyponatremia, the patient was studied for a suspected pan-hypopituitarism, which was confirmed on blood tests and explained by meningeal disease, in absence of disease localization in MR. Replacement treatment with levothyroxine, oral corticosteroids and desmopressin was administered. After the first two cycles of chemotherapy the patient obtained a PR and the unrelated donor research was started (having a brother suffered from a testicular cancer) with the intent of giving an early treatment with allogenic stem cells transplantation. The patient was discharged from the hospital in good clinical conditions. The next day, he came to our emergency room for unexplained episodes of seizures, confusion associated with urinary incontinence. The head CT and MR showed a hygroma with early signs of tonsillar herniation caused by liquor hypotension syndrome and he was treated with mannitol, dexamethasone and diuretics. In August, after completing first course A of R-Hyper-CVAD (CTX 300 mg/12 hours from day 1 to 3, doxorubicin 50 mg on day 4 and dexamethasone 40 mg from day 1 to 4 and from day 11 to 14), avoiding vincristine due to its neurotoxicity, the patient was again discharged in good clinical condition. After a couple of weeks, the patient was hospitalized again for FUO, headache and nausea. Antibiotics were started, with no microbiological finding. The patient underwent a new disease evaluation in PET-TC that showed a progressive disease (PD) in the same localizations that were known at diagnosis. New cutaneous lesions appeared that were consistent with BCP-ALL, while the hygroma was stable in MR and bone marrow aspirate was again negative for infiltrate. After one more week, the patient presented right eyelid ptosis (CT head negative) that was treated with no benefit with high dose dexamethasone and never resolved. The patient underwent second line treatment with the two cycles of HAM regimen (citarabine 3 g/mq every 12h from day 1 to day 4, mitoxantrone 10 mg/mq from day 3 to day 5). During this period, he suffered from septic shock caused by Klebsiella pneumonia carbapenemase-producing (KPC) and started on isavuconazole due to positivity of galactomannan (GMN) antigen in samples of bronchoalveolar lavage and fungus ball images at CT scan. On day 14 of the second cycle of HAM, the patient presented increasingly elevated lactate dehyogenase (LDH) levels, a growth of the cutaneous lesions and was evaluated with a total body CT scan that showed PD and bilateral thrombosis of both pulmonary arteries, treated with anticoagulant therapy. In October after the clinical progression of disease, the patient began third line therapy with “Teddi” [ 3] regimen (temozolomide 200 mg os from day 1 to day 5, etoposide 100 mg/mq from day 1 to day 3, dexamethasone 40 mg from day 1 to day 4, caelyx 40 mg/mq on day 1 and daily ibrutinib 560 mg die os). On day 15, the patient suffered from severe hyponatremia, LDH levels were at highest and he suffered from episodes of unconsciousness. On day 16, ibrutinib was suspended due to a severe hemorrhagic cystitis caused by KPC. On day 24, the patient suffered from one more epilepsy attack and, sequentially, a cardio-respiratory arrest. Despite reanimation, the patient died after three more days.
Discussion
The presence of both C-Myc and Bcl-2 rearrangements in B-cell lymphomas defines a DHI: such finding is associated with a higher CNS involvement, a more advanced stage, an inferior response to chemotherapy and a poorer prognosis [ 4]. DLBCLs harbor simultaneous Myc and Bcl-2/6 rearrangements in at least 5-10% of cases [ 5]. A single rearrangement of Myc occurs in 5-10% of BCP-ALL, is associated with hypodiploidy and mutated TP53 and brings a poorer outcome [ 6, 7]. Bcl-2 is mostly rearranged in proximity of IgH locus, while Myc is rearranged in both IgH – non IG locus, with no difference in survival. DHI with Bcl-6 is less common [ 8]. Miyaoka et al. reviewed 25 patients with this particular finding in follicular lymphomas, with a conclusion that there were no prognostic differences instead of HGBLs [ 9]. De novo DH-BCP-ALLs affect patients of all ages, with no significant difference in gender; usually present themselves with high WBC counts, elevated LDH levels and systemic symptoms. Most cases show a diffuse bone marrow infiltration, CNS involvement (one/third showed meningeal infiltration) and extra-medullary disease, which is mostly gastrointestinal, in the salivary glands and paraosseal. Immuno-phenotypic analysis shows immature B-cell onset (CD10+, CD19+, TdT+, IgM-), high expression of Myc e Bcl-2 and a complex karyotype. In the previously cited review, 60% of the patients, which were enrolled in different protocols (mainly ALL-based), achieved nearly hematologic remission. All patients received an intrathecal prophylaxis with methotrexate and cytarabine; however, most of them relapsed within 12 months with a median OS after relapse of 2.5 months. In a 2015 review, Wei Liu et al. presented three pediatric cases of DH-BCP-ALLs that shared the same clinical and pathological characteristic previously described for adult patients [ 10].
Conclusion
Our patient shared clinical features with those known in literature, such as the CNS involvement, as previously reported in DH-BCP-ALLs reviews, but differed in immune-phenotypical analysis (IgM positive, Tdt+ in 10% of the cells), in cytogenetic (normal karyotype) and in the absence of disease infiltration in bone marrow. This case report confirms that DHI and THI do not fall into a single WHO category, as Myc and Bcl-2 rearrangements are observed in DLBCL, in FL and also in BCP-ALL. We can infer that, when DH-BCP-ALLs diagnosis is made, intensive treatment regimens are required to achieve an early complete response, however there is no evidence of which regimen should be preferred. Early relapse is the main factor correlating to a poor prognosis in all cases observed so a consolidation therapy, such as allogenic stem cell transplantation, could be considered. Screening search of Myc, Bcl-2 and Bcl-6 may be helpful to recognize de novo DH-BCP-ALLs and provide a fine therapeutic approach to such disease. Flow cytometry plays a crucial role, analysis of CD3, CD10, CD13, CD33, CD19, CD20, surface kappa/lambda light chain, cytoplasmic TdT and cytoplasmic CD 22 is recommended.
Contributors: RC, FM: manuscript writing, literature review and patient management; FE, AZ: reviewing the literature and manuscript editing; VMR, IP, MR: critical inputs into the manuscript. LA, MC: reviewing the literature. RC will act as a study guarantor. All authors approved the final version of this manuscript. Funding: None; Competing interests: None stated.
References - Bischin AM, Dorer R, Aboulafia DM. Transformation of follicular lymphoma to a high-grade B-cell lymphoma with MYC and BCL2 translocations and overlapping features of Burkitt lymphoma and acute lymphoblastic leukemia: A case report and literature review. Clin Med Insights. Blood Disord. 2017;28:10.
- Ferreri AJM, Holdhoff M, Nayak L, Rubenstein JL. Evolving treatments for primary central nervous system lymphoma. Am Soc Clin Oncol Educ Book. 2019;39:454-466.
- Lionakis MS, Dunleavy K, Roschewski M, Widemann Brigitte C, Butman John A, Schmitz R, et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell. 2017;31:833-843.
- Niitsu N, Okamoto M, Miura I, Hirano M. Clinical features and prognosis of de novo diffuse large B-cell lymphoma with t(14;18) and 8q24/c-MYC translocations. Leukemia. 2009;23:777-783.
- Adam MP, Nabhan C, Smith SM. MYC-associated and Double-hit lymphomas: A review of pathobiology, prognosis, and therapeutic approaches. Cancer. 2014;120:2884-3895.
- Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A, et al. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014;124:251-258.
- Kelemen K, Holden J, Johnson L, Davion S. Immunophenotypic and cytogenetic findings of B-lymphoblastic leukemia/lymphoma associated with combined IgH/BCL2 and MYC rearrangement. Cytometry B Clin Cytom. 2017;92:310-314.
- Aukema Sietse M, Siebert R, Ed Schuuring, Imhoff W Van, Kluins-Nelemans C, Boerma J, et al. Double-hit B-Cell lymphomas. Blood. 2011;117:2319-2331.
- Miyaoka M, Kikuti Yara Y, Carreras J, Ikoma H, Hiraiwa S, Ichiki A, et al. Clinicopathological and genomic analysis of Double-hit follicular lymphoma: comparison with high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Modern Pathology. 2015;31:313-326.
- L Wei, Shimin Hu, M Konopleva, Joseph D. Khoury, N Kalhor, et al. De Novo MYC and BCL2 Double-hit B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in pediatric and young adult patients associated with poor prognosis. Pediatric Hematology and Oncology. 2015;32:535-547.
|