|
Luis Alberto Aparicio Vera, Jessica Mariana Suárez Martínez Pediatric Rheumatology Department, Hospital para el niño Poblano, Boulevard del Niño Poblano 5307, Concepción de la Cruz, 7219, San Andrés Cholula, Puebla.
Corresponding Author:
Dr Jessica M. Suárez Martínez Email: jesisu92@hotmail.com
Abstract
Background: Budd-Chiari syndrome is a rare entity in childhood. Antiphospholipid syndrome is an autoimmune disease characterized by the presence of thrombosis or obstetrical complications. The presentation of both diseases is rare. Currently there are few reports in the literature of this association. Case Report: A 15-year-old male patient presented with generalized pallor, Raynaud phenomenon and pain abdomen. Imaging studies reveal portal vein and splenic vein thrombosis corroborating with angiography. Based on the thrombosis presentation, Raynaud phenomenon, aTTP prolongation, thrombocytopenia and complement consumption, diagnosis of antiphospholipid syndrome was made. He continued treatment with anticoagulation, in addition to steroid and azathioprine, presenting adequate clinical evolution. Conclusion: The seronegative antiphospholipid syndrome represents a diagnostic challenge. It´s essential to maintain a suspected diagnosis of antiphospholipid syndrome as a cause of Budd-Chiari syndrome, even in it´s seronegative presentation.
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffa40a370000009706000001000300 6go6ckt5b5idvals|3111 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Budd-Chiari syndrome (BCS) is a result of impaired hepatic venous outflow at any point from the efferent acinar vein up to the end of the inferior vena cava. Primary BCS occurs in patients with primary hematological disorders or hypercoagulable conditions [ 1]. Antiphospholipid syndrome is a systemic autoimmune disease characterized by thrombotic events in patients with antiphospholipid antibodies [ 2]. These autoantibodies include anticardiolipin antibodies, anti-ß2-glycoprotein 1 antibodies and lupus anticoagulant. The pathophysiological hallmark is thrombosis, but other factors such as complement activation might be important [ 3]. The diagnosis of APS is based on the combination of clinical features (for example, thrombosis in the arteries, veins and/or small vessels or obstetrical complications such as recurrent miscarriage and placental insufficiency) and the detection of circulating antiphospholipid antibodies [ 3]. The cases with APS seronegative are the patients with migraine, stroke, several previous miscarriages, thrombocytopenia, livedo reticularis and thrombosis whose antiphospholipid tests are doggedly negative [ 4]. Darwish and collaborators in 2009 founded that 84% of patients with BCS have at least one thrombophilic disorder and that 74% of these same patients had more than one prothrombotic condition, myeloproliferative disease in most cases [ 5]. Here we present the case of an adolescent, who comes to our institution for a condition characterized by constitutional symptoms, gastrointestinal symptoms, Raynaud phenomenon, bleeding disorders, a diagnosis of BCS subacute form is integrated by clinical presentation with bleeding secondary to esophageal varices and imaging studies. This is a high impact clinical case, since both etiologies are infrequent, and there is little evidence of relationship between them.
Case Report
The patient was a 15-year-old male who came to our hospital with history of sporadic epistaxis that remitted spontaneously, received prior treatment with ferrous fumarate and vitamin K without improvement. He underwent turbinate cauterization four months prior to arrival at our hospital. Two months before presentation, he had hyporexia, weight loss of 11.5 kg, accompanied by abdominal pain beginning in the epigastrium, later radiating to the left hypochondrium and lumbar region of the same side. Diarrheal evacuations of one month of evolution were treated with cephalexin without improvement, and vomiting of biliary content occurred on several occasions’ days prior to admission. At his arrival he was found with pallor, Raynaud's phenomenon, painful abdomen on median and deep palpation in the right hypochondrium, positive Murphy's sign, hepatic border 3 cm below the costal margin. Laboratory studies were requested and a TTP prolongation was found [Table 1]. The abdominal ultrasound detected portal vein dilatation 23 mm, with presence of dense echoes due to thrombi and absence of vascular flow, portal and splenic vein with thrombi inside [Fig.1,2]. Abdominal CT angiography reported filling defect in portal vein with 68% occupancy in the distal third, with apparent contrast intercalated passage; occlusion of 45.7% in proximal third. the portal vein caliber was 19.9 mm [Fig.3]. A diagnosis of portal and mesenteric thrombosis was made with imaging studies and signs of portal hypertension. The treatment was started with enoxaparin. The patient developed hypovolemic shock after 10 days of hospitalization secondary to upper gastrointestinal tract hemorrhage, presenting three melenic evacuations; requiring vasopressor support and mechanical ventilation support for 3 days. Diagnostic endoscopy was performed, finding grade 2 esophageal varices and grade 3 hypertensive gastropathy.
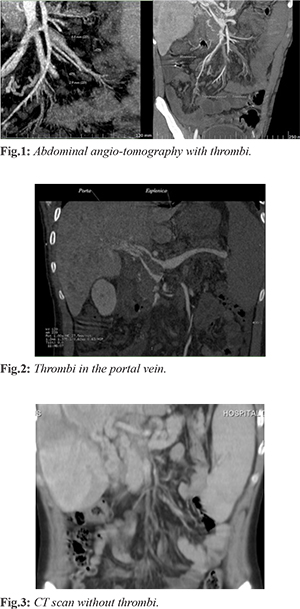
A diagnosis of Budd Chiari subacute presentation was made due to abdominal pain, hepatomegaly, imaging studies with portal and mesenteric vein thrombosis, portal hypertension and secondary esophageal varices. An etiological approach was initiated, ruling out infectious, metabolic and primary coagulation defect associated etiologies. In the rheumatologic approach, a conventional and enlarged negative antiphospholipid antibody profile was reported. The diagnosis of sero-negative antiphospholipid syndrome was made base on Raynaud phenomenon, aTTP prolongation, thrombocytopenia, complement decrease, portal and mesenteric thrombosis, and negative approach for some other differential diagnosis [Table 1]. He continued treatment with enoxaparin, in addition to steroid and azathioprine, presenting adequate clinical evolution. One year after beginning treatment a new abdominal CT was made demonstrating absence of thrombosis. He is currently under surveillance [Fig.3].
Discussion
Budd-Chiari syndrome (BCS) groups different pathological processes that are characterized by the obstruction of hepatic venous ouflow which may occur from the hepatic venules, hepatic veins and the inferior vena cava up to the right atrium after excluding liver parenchyma and cardiac causes [ 6]. The probable associated etiologies include prothrombotic states, oncological, infectious, autoimmune or intrinsic coagulation defects. BCS can be observed at any age, but is uncommon in children and only 5% were found to be below 12 years of age [ 7]. The diagnosis is established by demonstrating obstruction of the venous outflow and structural changes of the liver, portal or venous system. Laboratory and hematological tests are an integral part of the comprehensive workup and are invaluable in recognizing coagulation disorders [ 8]. In the case of our patient, the diagnosis of Bud-Chiari syndrome was based on the presence of constitutional symptoms, abdominal pain, vomiting, and hepatomegaly. It was later corroborated with the obstruction of the hepatic venous flow observed in imaging studies. Once the diagnosis of Bud-Chiari syndrome has been established, the next challenge corresponds to the etiological approach. In the case or our patient, the approach mainly included infectious, gastrointestinal and hematological causes [Table 1]. Antiphospholipid syndrome (APS) is an autoimmune disease characterized by the presence of antiphospholipid antibodies, plus a variety of clinical phenotypes including thrombosis and non-thrombotic manifestations [ 3]. The cardinal antibodies related with APS are the anti-ß2-glycoprotein 1 antibodies, anticardiolipin antibodies and lupus anticoagulant. The diagnosis is made based on the Sydney criteria. This classification criteria for antiphospholipid syndrome are referred as the Miyakis criteria [ 9]. In addition to the aforementioned, there is a group of patients whose disease has a clear association with autoimmunity but who persistently show negativity for autoantibodies. This group of diseases has been generically called “seronegative diseases” [ 10]. The diagnosis in this group of patients represents a challenge, and the underdiagnosis of this disease can be associated with high mortality due to the risk of thrombosis in vital organs. In addition to all this, we can find other clinical data of APS that do not belong to the diagnostic criteria of the disease, making the diagnosis even more difficult [4]. The diagnosis of APS in our patient was based on the presence of thrombosis, thrombocytopenia, Raynaud´s phenomenon and the ruling out of other possible causes. However, our patient persistently presented negativity for aPS, which in previous studies has been related to different causes, including the “consumption” of these antibodies during the acute thrombotic process [ 11]. The presence of Raynaud´s phenomenon and thrombocytopenia have been described as non-thrombotic symptoms of APS, or “extra criteria”, referring to their absence in the current criteria for the diagnosis of the disease [ 12]. Currently, it has been suggested by many experts to include these non-thrombotic symptoms in the criteria for classifying the disease based on the wide variety of symptoms that can occur in the disease. The link between APS and BCS has been previously described, mainly in the adult population and to a lesser extent in the pediatric population. In some cases, it has been described as primary APS and in other cases associated with the onset or evolution of lupus [ 13, 14]. In 2001, Espinosa et al, reported 43 patients on Budd-Chiari syndrome secondary to antiphospholipid syndrome, including 6 pediatric patients, all of them reported with primary APS, the youngest being 2 years of age [ 15]. It is relevant that a patient reported a PS negative during the height of her BCS, but positive 6 years later. The most prevalent APS was anti-cardiolipin antibody. In 2005, Carbajal et al, carried out a retrospective study which included patients with APS and its relationship with BCS. Seven (29%) pediatric patients with APS and BCS were reported in this study. The age of presentation was 10 to 15 years. A slight predominance of the female gender was reported with 4 of the 7 cases. All 7 patients had portal thrombosis. 6 patients were reported as primary APS, and all the patients had positive anti-cardiolipin antibodies and lupus anticoagulant [ 16]. The clinical phenotype of our patient coincides with the age reported in this study, even with the less frequent symptoms. Our patient also coincides with the serological phenotype at the level of liver enzymes and also with the predominance of portal thrombosis. It is important to note that no patient with suspected seronegative APS was reported, as well as non-thrombotic data on APS. In recent years, several case reports have been published in adult population, some associated with specific situations such as liver transplantation, Libman-Sacks endocarditis or relapsing polychondritis [ 17- 21]. In some cases, multiple prothrombotic factors may be associated. Among thrombophilic states, primary myeloproliferative disorders are rare in children. However, other prothrombotic conditions should be considered, including inflammatory bowel disease, paroxysmal nocturnal hemoglobinuria, hyper-homocysteinemia, inherited prothrombotic disorders such as protein C, S, antithrombin III deficiencies and factor V Leiden mutation and methylenetetrahydrofolate reductase gene mutation. These diseases usually present in the absence of associated liver disease [ 22, 23]. In the case of our patient this approach was carried out in a staggered manner, reasonably ruling out these etiologies [Table 1]. In addition to the previous approach, other causes of rheumatological disease associated with thrombosis were also considered, such as systemic lupus erythematosus and Behcet´s disease. However, it was not possible to integrate these diagnosis in the context of our patient [ 24]. Treatment in these patients is aimed at reducing the risk of subsequent thrombosis. Hydroxychloroquine has shown an important role in the treatment of patients with APS under certain conditions, so it´s use can be reasonably considered. After thrombosis, anticoagulation with low molecular weight heparin is considered the treatment of choice [ 25, 26]. In the case of our patient, a good response was obtained with treatment based on hydroxychloroquine, low molecular weight heparin, prednisone, and azathioprine. The latter were considered since they have been described as useful in patients with hematological symptoms associated withs APS, such as thrombocytopenia, as was our patient [ 27]. BCS treatment must be multidisciplinary. The management decision depends on several factors including local expertise, presentation of patient, site of blockage and structural anatomy. In the present era, most of the BCS are managed with radiological intervention [ 28], however, the risk of endothelial damage and the of a new thrombosis was a limitation for this therapeutic option in our patient. Prothrombotic states need to be worked up carefully. Children with BCS, including those with prothrombotic disorders as the primary etiology of the disease can have a good long-term outcome with a multidisciplinary approach. At present, our patient continues under surveillance, with control imaging studies where the absence of thrombosis is documented.
Conclusion
The diagnostic approach to BCS must be performed thoroughly. It´s association with prothrombotic states has been well established. The diagnosis of APS in pediatrics can be a real challenge for the clinician and even more so for the pediatric rheumatologist. Its consideration as a differential diagnosis is very important, including its sero-negative varieties, and those with non-thrombotic presentation. Its timely diagnosis and treatment can result in a significant decrease in morbidity and mortality. Our case corresponds to the first case reported in the literature of seronegative APS with BCS.
Contributors: LAAV: manuscript editing, patient management; JMSM: manuscript writing, patient management. JMSM will act as a study guarantor. Both authors approved the final version of this manuscript and are responsible for all aspects of this study. Funding: None; Competing interests: None stated.
References
- Grus T, Lambert L, Grusova G, Banerjee R, Burgetova A. Budd-Chiari syndrome. Prague Medical Report. 2017;118:69-80.
- Lirola M, Camacho M. Síndrome antifosfolípido. Protoc diagn ter Pediatr. 2020;2:141-154.
- Scrhebier K, Sciascia S, de Groot P, Devreese K, Jacobsen S, Ruiz-Irastroza G. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4:19.
- Hughes GRV, Khamashta MA. Seronegative antiphospholipid syndrome: an update. Lupus. 2019;28:273-274.
- Correa GS, Ramírez AC, Espinoza HY, Espinoza HYP, Restrepo GJC. Síndrome de Budd Chiari: revisión de tema. Rev Col Gastroenterol. 2016;31(3):242-252.
- Coilly A, Potier P, Broué P, Kounis I, Valla D, Hillaire S. Budd-Chiari syndrome. Clin Res Hepatol Gastroenterol. 2020;44(4):420-425.
- Misra V, Verma K, Kumar SD, Misra SP. The Budd-Chiari syndrome in a child: A case report and review of the literature. J Clin Diagn Res. 2012;6(10):1783-1785.
- Grus T, Lambert L, Grusová G, Banerjee R, Burgetová A. Budd-Chiari syndrome. Prague Medical Report. 2017;118;69-80.
- Miyakis, S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306.
- Sciascia S, Amigo MC, Roccatello D, Khamashta M. Diagnosing antiphospholipid syndrome: “extra-criteria” manifestations and technical advances. Nat Rev Rheumatol. 2017;13:548-560.
- Hughes GRV, Khamashta MA. Seronegative antiphospholipid syndrome. Ann Rheum Dis. 2003;62:1127.
- Pignatelli P, Ettorre E, Menichelli D, Pani A, Violi F, Pastori D. Seronegative antiphospholipid syndrome refining the value of “non-criteria” antibodies for diagnosis and clinical management. Haematologica. 2020;105(3):562-572.
- Pelletier S, Landi B, Piette JC, Ekert P, Coutellier A, Desmoulins C, et al. Antiphospholipid syndrome as the second cause of non-tumorous Budd-Chiari syndrome. Journal of Hepatology. 1994;21:76-80.
- Pandiaraja J, Sathyaseelan A. Budd-Chiari syndrome as an initial manifestation of systemic lupus erythematosus. Journal of Clinical and Diagnostic Research. 2016;104(4):1-2.
- Espinosa G, Font J, García PJC, Tàssies D, Reverter JC, Gaig C. Budd-Chiari syndrome secondary to antiphospholipid syndrome. Medicine (Baltimore). 2001;80(6):345-354.
- Carbajal RL, Vargas QE, Ramírez MJA. Síndrome de Budd-Chiari (SBC) y su asociación con síndrome antifosfolípidos. Acta Pediatr Mex. 2005;26(6):302-307.
- Lastra CG, Soriano RJ, Carrera GE, Guevara LB, Durán PM. Síndrome de Budd-Chiari secundario a síndrome de anticuerpos antifosfolípidos primario. Reporte de un caso y revisión de la literatura. Rev Med Hosp Gen Mex. 2002;65(3):164-167.
- Chinen N, Koyama Y, Sato S, Susuki Y. A case of acute Budd-Chiari syndrome complicating primary antiphospholipid syndrome presenting as acuteabdomen and responding to tight anticoagulant therapy. Case Reports in Rheumatology. 2016:9565427.
- Obed. A, Bashir A, Jarrad A. A case of live donor liver transplantation in acute-on-chronic liver failure with Budd Chiari syndrome: donor and recipient with antiphospholipid antibody syndrome. Am J Case Rep. 2018;19:767-772.
- Goldhar HA, O’Meara P, Castelluci LA. A case of antiphospholipid syndrome presenting cryptogenically as Budd-Chiari syndrome, then fulminantly as Libman-Sacks endocarditis. BJM Case Rep. 2019;12:1-3.
- Sebastiani M, Manzini CU, Campomori F, Spinella A, Vacchi C, Giuggoli D, et al. Unusual association between Budd-Chiari syndrome secondary to antiphospholipid syndrome and relapsing polychondritis: a case report and review of the literature. Clin Rheumatol. 2013;32:905-907.
- Yogesh KC, Bodh V. Portal vein thrombosis. J Clin Exp Hepatol. 2015;5(1):22-40.
- Fousekis FS, Theopistos VI, Katsanos KH, Tsianos EV, Christodoulou DK. Hepatobiliary manifestations and complications in inflammatory bowel disease: A review. Gastroenterology Res. 2018;11(2):83-94.
- Emmi G, Bettiol A, Silvestri E, Di Scala G, Becatti M, Fiorillo C. Vascular Behcet´s syndrome: an update. Internal and Emergency Medicine. 2019;14:645-652.
- Auguiar CL, Soybilgic A, Avcin T, Myones BL. Pediatric antiphospholipid syndrome. Curr Rheumatol Rep. 2015;17(4):27.
- Dobrowolski C, Erkan D. Treatment of antiphospholipid syndrome beyond anticoagulation. Clinical Immunol. 2019;206:53-62.
- Negrini S, Pappalardo F, Murdaca G, Indiveri F, Puppo F. The antiphospholipid syndrome: from pathophysiology to treatment. Clin Exp Med. 2016;17(3):257-267.
- Nobre S, Khanna R, Bab N, Kyrana E. Primary Budd-Chiari syndrome in children: King´s College Hospital Experience. J Pediatr Gastroenterol Nutr. 2017;65:93-96.
|