|
Dimitrios Rallis1, Maria Baltogianni1, Evita Evangelia Christou2, Paraskevas Zafeiropoulos2, Nikolaos Marinakis3, Joanne Traeger-Synodinou3, Ioannis Asproudis2, Georgios Makrydimas4, Vasileios Giapros1 1Neonatal Intensive Care Unit, University of Ioannina, Faculty of Medicine, Ioannina, Greece; 2University Ophthalmology Clinic, University of Ioannina, Faculty of Medicine, Ioannina, Greece; 3Laboratory of Medical Genetics, National and Kapodistrian University of Athens, St Sophia's Children Hospital, Athens, Greece; 4Department of Obstetrics and Gynaecology, University of Ioannina, Faculty of Medicine, Ioannina, Greece.
Corresponding Author:
Dr Dimitrios Rallis Email: drallis@uoi.gr
Abstract
Background: Noonan syndrome (NS) is a genetically heterogeneous disorder, with several causative variants identified. Up to date, novel leucine zipper-like transcriptional regulator-1 (LZTR1) variants have been reported in a few cases with Noonan syndrome. Case Report: We report the case of a male infant with the phenotypic characteristics of Noonan syndrome, unusual ocular findings, and a LZTR1 variant. Our patient was born prematurely, with antenatal diagnosis of non-immune hydrops, and unremarkable amniocentesis for known Noonan variations. He had dysmorphic features (low set ears, wide nasal bridge, undescended testes), normal weight, and length. After one month of age he developed pulmonary stenosis, and significant extrauterine growth restriction, whereas ocular examination revealed retinal vascular tortuosity. He was discharged at seven months of corrected gestational age, on nocturnal low flow oxygen. Based on van der Burgt’s criteria the diagnosis of NS was established. Whole exome sequence analysis revealed a heterozygous variant in the LZTR1 gene (NM_006767.4):c.27delG (p.(Gln10Argfs*15). Conclusion: Given that new LZTR1 mutations may be identified, the evaluation of an extended genetic panel should be applied in suspected NS cases.
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ffa404380000009d06000001000700 6go6ckt5b5idvals|3124 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
Noonan syndrome (NS) (MIM#163950) is a genetic disorder belonging to RASopathies, which are characterized by the deregulation of the RAS/mitogen-activated protein kinase (MAPK) pathway [ 1]. The prevalence of NS is estimated between 1:1000 to 1:2500 live births [ 2]. Antenatally, NS can be associated with cystic hygroma, nuchal lucency, or hydrops fetalis, whereas postnatally, the clinical findings include distinctive facial dysmorphic features, congenital heart defects, and neck webbing [ 2]. Relatively common features are also clotting abnormalities, growth restriction, skeletal anomalies, lymphatic dysplasia’s, cryptorchidism, and cognitive deficits; however, in many cases the clinical characteristics may not be present at birth [ 2, 3]. In suspected cases, a genetic screening is essential to formulate the diagnosis. In nearly 75% of individuals with NS, mutations in PTPN11, SOS1, KRAS, NRAS, RAF1, BRAF, or MAP2K1 are detected [ 2]. Recently, novel variants have been identified including mutations affecting the leucine zipper-like transcriptional regulator-1 ( LZTR1) gene [ 4]. Of note, the causative association of LZTR1 mutations with the NS phenotype remains obscure, since some variants have been also reported in asymptomatic individuals [ 5]. In the current study, we report the case of a male infant with latter clinical manifestations of NS, unusual ocular findings, and a rare detection of a de novo heterozygous dominant LZTR1 mutation.
Case Report
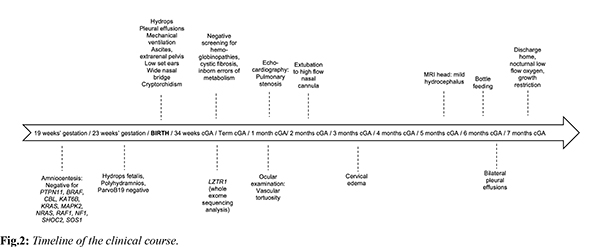
A male infant of non-consanguineous parents was born by caesarean section at 30+6 weeks, with a birth weight of 1950 g (90-95th centile), head circumference 28 cm (50th centile) and length 41 cm (97th centile), and the antenatal detection of non-immune hydrops fetalis. Amniocentesis had revealed a normal karyotype, whereas screening for PTPN11, BRAF, CBL, KAT6B, KRAS, MAPK2, NRAS, RAF1, NF1, SHOC2, SOS1 mutations, and maternal serology, including parvovirus B19, was unremarkable. Our patient required intubation after birth, commencing thereafter on mechanical ventilation. He was edematous, with low set ears, wide nasal bridge, and undescended testes [Fig.1a]. Chest radiology revealed bilateral pleural effusions requiring chest draining. Echocardiography on the first day of age was unremarkable, whereas abdominal ultrasound revealed mild ascites and extrarenal pelvis. Extensive laboratory investigations were unremarkable, including full blood count and microscopy, urea and electrolytes, liver function tests, lipid and bone profiles, thyroid function tests, clotting, screening for hemoglobinopathies, glucose-6-phosphate dehydrogenase deficiency, cystic fibrosis, intrauterine infections, or metabolic diseases. In view of the diagnostic dilemmas, we performed a proband only whole exome sequencing (WES), applying a large bioinformatic panel of phenotypic associated genes. Bioinformatic algorithms were utilized for the detection of the variant in comparison to the GRCh37/hg19 reference gene. An heterozygous variant (NM_006767.4):c.27delG, p.(Gln10Argfs*15) was identified in the LZTR1 gene. According to multiple database analyses (VarSome, gnomAD, UMD-predictor, Shift, Polyphen, etc.), we concluded that the LZTR1 was a pathogenic variant. Family segregation using targeted Sanger sequencing revealed the de novo origin of the variant; paternity confirmation was also performed.
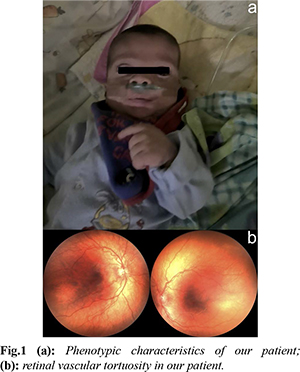
At one month of corrected age, our patient developed mild pulmonary stenosis. Furthermore, brain ultrasound showed mild ventricular dilation, whereas magnetic resonance imaging (MRI) revealed mild hydrocephalus; interestingly, ocular examination revealed significant retinal vascular tortuosity [Fig.1b]. During the first months of age, he presented episodes of cervical edema with spontaneous resolution; however, no evidence of cystic collections or lymphatic ectasias was found in MRI. Overall, our patient was ventilated for 86 days, with gradual de-escalation until discharge to nocturnal low flow oxygen. He remained tube-fed until six months of corrected age when he managed to tolerate bottle-feeding; however, he suffered a significant extrauterine growth restriction with body weight and length below the 3rd centile. Based on the non-typical face dysmorphology, the cryptorchidism, and the latter developed pulmonary valve stenosis and the significant growth restriction, the diagnosis of NS was established based on van der Burgt’s criteria [3]. On discharge the infant had mild pleural effusions, and he was mildly hypotonic, with normal cognitive and motor function for his age. The timeline of his clinical course is depicted in Fig.2.
Discussion
In the current case, we reported an infant with the phenotypic diagnosis of NS presented later during the first months of age. In many cases the diagnosis of NS may be problematic, given the wide phenotypic variability of the disease and that many clinical aspects may not be present at birth and become apparent with age [ 2]. Infants usually present normal stature followed by extrauterine growth restriction, whereas facial dysmorphism and pectus deformities become more obvious later in life [ 2]. Cardiac defects may appear during the first year of life and in addition to short stature, are considered the major causes for patients with NS requiring medical attention. Notably, our patient presented with non-immune hydrops and limited facial features at birth, whereas the cardiac, lymphatic, and growth manifestations appeared later during infancy. Interestingly, although a wide spectrum of ocular manifestations has been described in NS, limited data have been reported regarding vascular tortuosity [ 6]. Vascular tortuosity has been reported as a rare ocular finding in syndromic patients with congenital heart diseases, that could be attributed to hypoxaemia and cyanosis [ 7]. In our case, given that no evident of intracranial or clotting abnormality existed, we speculate that the prolonged period of mechanical ventilation and events of hypoxaemia may have led to the manifestations of vascular tortuosity. The application of an extended genetic panel including novel variants such as LZTR1 has aid to the detection of previously undetected NS cases. The mechanism by which variants in LZTR1 confer an NS phenotype is characterized by a dysregulation of the RAS/MAPK cascade with subsequent alterations in the expression of ERK [ 8]. Previous experimental studies have indicated the implication of LZTR1 in the RAS/MAPK pathway, which leads to the inhibition of RAS activity [ 9]. Furthermore, LZTR1 has been also associated with the RAF1/SHOC2/PPP1CB complex, which is responsible for regulating the RAS/MAPK pathway, and that has been implicated in the pathogenesis of NS [ 10]. Evidence suggested that LZTR1 plays a negative modulatory role in controlling RAS function and MAPK signaling; dominant NS-causing LZTR1 mutations likely impair proper binding to RAS substrates, resulting in RAS/MAPK signaling upregulation, whereas missense LZTR1 variants associated with recessive NS behave as loss-of-function mutations in the context of RAS/MAPK signaling [ 11]. In our case, a rare de novo heterozygous dominant LZTR1 mutation associated with NS was detected. A few cases have been reported so far regarding the association of variants in LZTR1 with NS [5,10,12-15]. Chen et al. detected LZTR1 variants in two amongst 27 patients with NS, although the authors did not consider them responsible for the clinical phenotype [ 12]. Moreover, Yamamoto et al. performing WES in 50 NS probands with previously uncharacterized pathogenic variants associated with NS, reported five LZTR1 variants predicted to cause NS [ 15]. Also, Guemes et al. reported three additional new patients with LZRT1 variants due to a different genetic transmission pattern responsible for NS [ 13], whereas Johnston et al. reported their findings in twelve families with an autosomal recessive inheritance of LZTR1-linked NS, with mutations spanning throughout the protein [ 5]. The authors, nevertheless, reported that some heterozygous parents showed mild signs of NS, indicating incomplete penetrance of LZTR1 [ 5]. In the same aspect, Umeki et al. identified eight LZTR1 variants, including a de novo variant, in seven probands that had not manifested the full clinical spectrum of NS [ 10].
Conclusion
We described the case of an individual with the phenotypic characteristics of NS and the detection of a previously identified mutation in LZTR1 implicated in the RAS/MAPK pathway. The evaluation of an extended genetic panel should be applied in suspected NS cases, in view of new LZTR1 mutations that may be identified.
Contributors: DR and EEC wrote the initial draft. MB, PZ, NM, JTS, IA, GM, and VG reviewed and revised the manuscript. DR will act as a study guarantor. All authors approved the final manuscript and agreed to be accountable for all aspects of the work. Funding: None; Competing interests: None stated. Abbreviations: cGA, corrected gestational age; LZTR1, leucine zipper-like transcriptional regulator-1; MRI, magnetic resonance imaging.
Statement of Ethics
Full informed consent was obtained from the infant’s mother for the genetic tests and reporting the clinical information and any accompanying images of the infant. The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008. The study protocol was reviewed and approved by the Ethics Committee of the Ioannina University Hospital, approval number 650/03-09-21. Data Availability Statement: The datasets generated during and/or analyzed during the current study are not publicly available due to patients’ confidentiality agreements but are available from the corresponding author on reasonable request.
References - Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61(1):33-39.
- Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab. 2011;25(1):161-179.
- van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007;2:4.
- El Bouchikhi I, Belhassan K, Moufid FZ, Iraqui Houssaini M, Bouguenouch L, Samri I, et al. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int J Pediatr Adolesc Med. 2016;3(4):133-142.
- Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med. 2018;20(10):1175-1185.
- Christou EE, Zafeiropoulos P, Rallis D, Baltogianni M, Asproudis C, Stefaniotou M, et al. A narrative review of the ocular manifestations in Noonan syndrome. Semin Ophthalmol. 2022;37:215-221.
- Mansour AM, Bitar FF, Traboulsi EI, Kassak KM, Obeid MY, Megarbane A, et al. Ocular pathology in congenital heart disease. Eye (Lond). 2005;19(1):29-34.
- Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1(1):2-26.
- Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science. 2018;362(6419):1177-1182.
- Umeki I, Niihori T, Abe T, Kanno SI, Okamoto N, Mizuno S, et al. Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1-PPP1CB complexes. Hum Genet. 2019;138(1):21-35.
- Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet. 2019;28(6):1007-1022.
- Chen PC, Yin J, Yu HW, Yuan T, Fernandez M, Yung CK, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci USA. 2014;111(31):11473-11478.
- Guemes M, Martin-Rivada A, Ortiz-Cabrera NV, Martos-Moreno GA, Pozo-Roman J, Argente J. LZTR1: Genotype expansion in Noonan syndrome. Horm Res Paediatr. 2019;92(4):269-275.
- Jacquinet A, Bonnard A, Capri Y, Martin D, Sadzot B, Bianchi E, et al. Oligo-astrocytoma in LZTR1-related Noonan syndrome. Eur J Med Genet. 2020;63(1):103617.
- Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52(6):413-421.
|