Introduction
Idiopathic inflammatory myopathies (IIMs) are rare group of systemic connective tissue diseases, which are characterized by symmetrical chronic inflammation and weakness of proximal muscles. Dermatomyositis (DM) is one of the IIMs and may occur in both adults and children. The incidence of DM is 9.63 cases per million population [
1].
The age of onset is between 5-18 years in juvenile DM. DM may present with skin eruptions which may be pruritic and scaly scalp or diffuse hair loss, in 40% of the cases. Muscle weakness may occur concurrently or after weeks to years [
2]. DM may be associated with systemic manifestations like malaise, arthralgia, dyspnoea, etc. However, subcutaneous calcifications are especially common in children [
3]. Here, we share an unusual case of DM, of a 5 year old boy who presented with urticarial skin rash and subcutaneous calcinosis.
Case Report
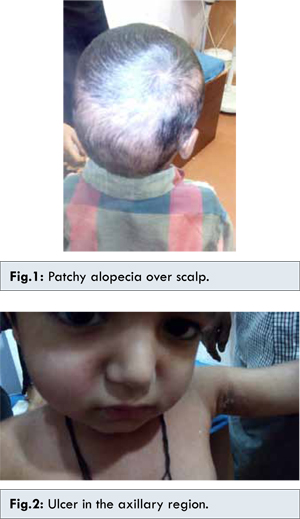
A five year old Indian boy presented with history of urticarial skin rash with itching over the limbs and the trunk since one year. Topical steroids were prescribed multiple times, but were ineffective and the rash continued to progress, both in extent and severity. He also had arthritis of small joints of hands and feet of more than one year duration. Since last one year, he also developed patchy alopecia over scalp [Fig.1]. Moreover, he was also suffering from non-healing ulcers over the elbow and the axillary region [Fig.2].
Blood count revealed reduced haemoglobin (9.9 gm/dL), and elevated ESR (110 mm/1 hour). His total WBC count was 15,200/cumm with neutrophilia and platelets were 4.9 lakhs/mm3. The peripheral smear showed thrombocytosis with neutrophilia and no evidence of abnormal cells. Rheumatoid factor (normal value 20-60 IU/mL) turned out to be negative (11 IU/mL) and ANA by ELISA was also negative. C-reactive protein (CRP) was high – 45 mg/L (normal value < 10 mg/L); lactate dehydrogenase, as expected, was high 550 U/L (normal value 105-333 U/L); creatine phosphokinase (CPK) and ferritin were within normal limits: 33 mcg/L (normal value 10-120 mcg/L) and 112 mcg/L (normal value 12-300 mcg/mL) respectively.
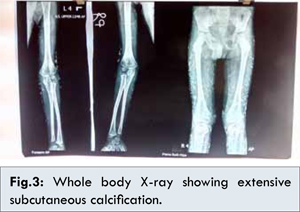
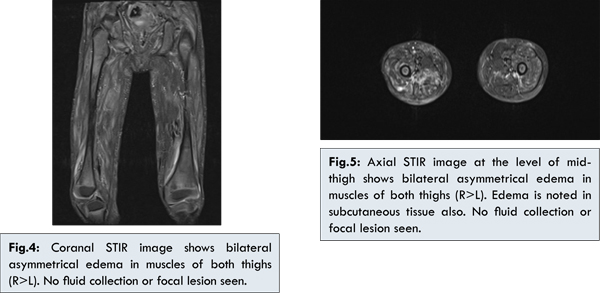
Since patient had nodular lesions, X ray of the whole body was done to look for calcinosis and was confirmed [Fig.3]. This finding was also confirmed by skin biopsy, which showed calcification. Now the suspicion of dermatomyositis was more evident and MRI of the thigh muscles was done [Fig 4,5] which showed myositis and a diagnosis of DM (JDM) was made.
Patient was administered methotrexate 5 mg/week initially for 2 months and then was maintained on 7.5 mg/week. Methyl prednisolone was initially started at 20 mg/day (OD) tapered to 2.5 mg (OD) taped over period of over 3 months later maintained on the same dose 2.5 mg every alternate day since 1 year. Axilla ulcer started healing within 6 weeks of starting the treatment and healed completely within 3 months. Skin rash disappeared within 3-4 months. After treatment, improvement was observed in haematological paramaeters, reduction in C-reactive protein, serum muscle enzyme levels (lactate dehydrogenase and creatine phosphokinase). Sign of healing appeared after 6 weeks and patient was in remission within 6 months and is maintained on the methotrexate 7.5 mg/week and methyl prednisolone 2.5 mg alternate day with calcium and vitamin D supplements. He is in remission since 1 year and on same treatment.
Discussion
The key to a favourable outcome in cases of JDM is early diagnosis and aggressive pharmacologic corticosteroid treatment [
4]. It is common for a patient to visit 4 to 5 doctors before the case is actually diagnosed. Subtle signs related to poor prognosis if missed, can lead to an unfavourable outcome. The delayed diagnosis can leave the case untreated and therefore is often accompanied by continued and increasing inflammation with potential systemic damage [
5].
The diagnostic criteria used to identify juvenile dermatomyositis includes characteristic skin rash, symmetrical muscle weakness of the upper and lower proximal muscles, increased levels of serum muscle enzymes, myopathic electromyography, or characteristic pathologic changes revealed by a muscle biopsy [
6]. It is important therefore to diagnose early so that corticosteroid therapy can be initiated early. Although corticosteroid therapy is the treatment mainstay, early introduction of steroid-sparing drugs like methotrexate often reduce the steroid burden [
7]. Alternatively, subcutaneous methotrexate can also be a feasible option and can reduce corticosteroid toxicity (weight gain, stunted growth, infections, hyperglycemia, cataract, etc.) [
8]. Methotrexate is capable of reducing disease activity and has also shown to reduce incidence of calcinosis [
7].
Other drugs which can act as steroid-sparing are hydroxychloroquine, cyclosporine, azathioprine and rituximab. In addition, physiotherapy and supplements like folic acid, vitamin D and calcium, regular use of sunscreen for the rash, help to improve the overall quality of life. Also, interdisciplinary care by various specialists, counselling of the family members and good compliance to the treatment plan optimize the outcome.
Most patients with DM survive. However, there may be residual weakness and contractures. The disease may unexpectedly remit in as many as 20% of the cases and on the other hand, about 5% of patients have a fulminant course with eventual death. Therefore, long-term therapy is warranted in most cases. The prognostic criteria contains persistent skin rash and cardiac or pulmonary involvement [
9].
Our patient has tolerated the drugs well but patient is short for his age. Methotrexate in adjunct to corticosteroid showed significant improvement on disease control, also methotrexate treatment leads to reduction in dose of corticosteroid which decreases the side effects of the corticosteroid therapy. Though the nodules which were calcifications still persist it is common with juvenile dermatomyositis even after different treatment option [
10].
It is important to note that muscle enzymes alone are poor indicators of the disease activity, and therefore must not be solely relied upon, magnetic resonance image is also a good diagnostic character. The most important marker of the disease activity in cases of DM is skin rash, even in presence of improvement in muscle weakness [
5]. As per new criteria for diagnosis of juvenile dermatomyositis, this case is probable juvenile dermatomyositis [Table 1].
Conclusion
Juvenile dermatomyositis is a rare disease which causes chronic disability in children. Early diagnosis and effective management with proper pharmacotherapy can prevent morbidity and mortality.
References
- Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146(1):26-30.
- Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. Often overlooked or misdiagnosed. JAMA. 1994;272(24):1939-1941.
- Na SJ, Kim SM, Sunwoo IN, Choi YC. Clinical characteristics and outcomes of juvenile and adult dermatomyositis. J Korean Med Sci. 2009;24(4):715-721.
- Pilkington CA, Wedderburn LR. Paediatric idiopathic inflammatory muscle disease: recognition and management. Drugs. 2005;65(10):1355-1365.
- Pachman LM, Abbott K, Dyer A, Lipton R, Ilowite N, et al. Duration of illness is an important variable for untreated children with juvenile dermatomyositis. J Pediatr. 2006;148(2):247-253.
- Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292(7):344-347.
- Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371(9631):2201-2212.
- Wedderburn LR, Rider LG. Juvenile dermatomyositis: new developments in pathogenesis, assessment and treatment. Best Pract Res Clin Rheumatol. 2009;23(5):665-678.
- Benbassat J, Gefel D, Larholt K, Sukenik S, Morgenstern V, Zlotnick A. Prognostic factors in polymyositis/dermatomyositis. A computer-assisted analysis of ninety-two cases. Arthritis Rheum. 1985;28:249-255.
- Patnaik S, Behera M, Mahakud N, Mohanty A. Juvenile Dermatomyositis with Calcinosis Cutis. Indian Journal of Clinical Practice. 2013;23(9):517-519.
- Murray KJ. Juvenile dermatomyositis: advances in understanding and management. APLAR J Rheumatol. 2003;6:50-63.