Introduction
There are various polyendocrine syndromes like Turner’s syndrome (chronic thyroiditis, type 1A diabetes), POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, serum M protein, skin changes), Type B insulin resistance (insulin-receptor autoantibodies, systemic lupus erythematosus) and autoimmune polyendocrine syndrome (APS I and II) [1]. Schmidt first described the relationship between lymphocytic thyroiditis and Addison’s and hence this combination is also referred to as Schmidt syndrome (APS II) [2]. We report a 27 year old lady with Schmidt syndrome who presented with primary infertility.
Case Report
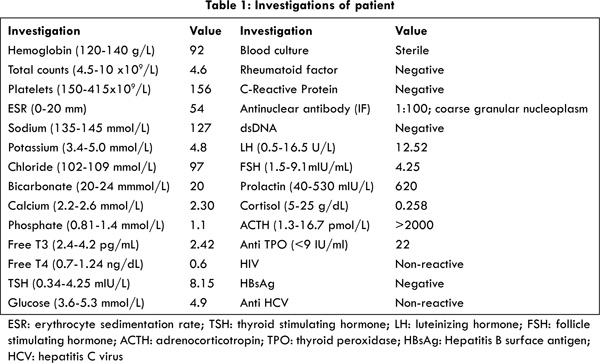
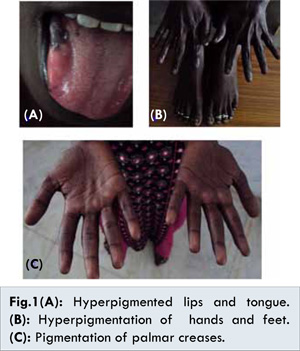
This lady had been married for the last 2 years and had been unable to conceive. She had history of secondary amenorrhea for the last one year. She had been evaluated by an obstetrician who had initiated her on L-thyroxine 50 µg once a day for subclinical hypothyroidism [Table 1]. Four weeks later she presented to our emergency department with dizziness and worsening fatigue. On examination she was conscious, alert, and thin-built with hyper-pigmentation of palms, soles, nail-beds and the tongue [Fig.1]. On questioning further, the woman had had fever, easy fatigability and polyarthralgias of 1½ months’ duration. It had been associated with loss of appetite and weight. Her husband had also noticed that her skin had become many shades darker during the previous 2-3 months. Her pulse was 88 beats/min, blood pressure (supine) 80/60 mm Hg, with normal respiration and systemic examination. Her investigations are listed in Table 1. She had normal liver and renal functions and abdominal ultrasonogram was normal. After obtaining samples, she was treated for presumed adrenocortical insufficiency, with intravenous hydrocortisone and dextrose-saline with which she significantly improved. Due to elevated ACTH, low cortisol, positive anti-TPO, she was continued on oral prednisolone, thyroxine and calcium. She was lost to follow-up after 2 months.
APS II (Schmidt syndrome) is a disorder with polygenic inheritance with an incidence about 1.4-4.5 per 100,000 population [2]. This is more common than type I and is seen from infancy through adulthood [1]. Most cases develop after the age of 20 years during the 3-4th decades [2,3]. Male female ratio is 1:3 [4]. It has autosomal dominant inheritance, with familial clustering and family members may manifest different diseases [3]. HLA-DQ2 and HLA-DQ8 are the alleles usually associated with APS [1]. When compared to the general population, patients with APS and Addison’s disease have 30-50 times increased risk of developing additional autoimmune diseases [1]. Carpenter’s syndrome includes the triad of type1 diabetes, Addison’s disease and autoimmune thyroid disease [2].
Addison’s disease is a rare component of APS and is usually symptomatic albeit diagnosed after many years [1]. The classic presentation is of hypotension due to adrenal insufficiency with intermittent episodes of hypoglycemia and fatigue [1]. Adrenocortical insufficiency is the initial abnormality in 50% of cases and is required for diagnosis [4]. Addison’s disease develops in stages, with metabolic parameters like elevated renin, reduced basal and stress cortisol levels developing prior to symptoms [1]. Diabetes usually heralds the syndrome in most cases [2]. Diabetes is observed in 20% of patients, while 10% of APSII have ovarian failure [4]. Frequency of individual diseases depends upon the population being studied [2].
Other component diseases may include vitiligo, hypergonadotropic hypogonadism, Graves’ disease, pernicious anaemia, alopecia, chronic thyroiditis, chronic atrophic gastritis and autoimmune chronic hepatitis [2]. APSIII includes thyroid autoimmunity and other autoimmunity [not related to diabetes/ Addison’s]. APS IV refers to other organ (=2) specific autoimmunity. These splitter groups [APS III and IV] are not universally agreed upon [1]. Increased pigmentation of not only normal skin, but also pre-existing naevi is observed patients with Addison’s disease in Schmidt syndrome [5]. Two patients have been reported with both connective tissue disease and APS [6], one of whom had cutaneous lupus. But our patient did not satisfy criteria for lupus apart from ANA positivity and alopecia. Autoantibodies in APS II include antibodies to thyroid peroxidase (TPO) and thyroglobulin (Hashimoto thyroiditis), TSH receptor and TPO [Graves’ disease], 21-hydroxylase, 17-hydroxylase and side chain cleavage enzyme (Addison’s disease), islet cells, GAD, IA2 and insulin (Type 1 diabetes) and many others [7]. Our hospital does not have facilities for these antibodies as well as testing for estrogen (E2). Decreased sodium, bicarbonate and chloride, increased potassium levels, and mild to moderate increased calcium level can be seen [4]. Hyponatremia is related to the presence of both hypothyroidism and hypoadrenalism and was seen in our patient [8]. Not ruling out coexisting adrenal disease in the previous hospital, resulted in adrenal crises after initiation of thyroxine.
Conclusion
Autoimmune polyendocrine syndromes can present to the obstetrics/gynecology department with infertility. Care needs to be taken to rule other coexisting conditions, since autoimmune antibodies can predict development of diseases even if asymptomatic at time of presentation [7]. Absence of antibodies does not exclude disease. Patients should be counseled regarding future risk of autoimmune diseases.
References
- Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. New Eng J Med. 2004;350:2068-2079.
- Betterle C, Lazzarotto F, Presotto F. Autoimmune polyglandular syndrome Type 2: the tip of an iceberg? Clin Exp Immunol. 2004;137:225-233.
- Kahaly GJ. Polyglandular autoimmune syndromes. Eur J Endocrinol. 2009;161:11-20.
- Majeroni BA, Patel P. Autoimmune polyglandular syndrome, type II. Am Fam Physician. 2007;75:667-670.
- Sunkel S, Wichmann-Hesse A, Gartner R, Hesse G. Increasing pigmentation in Schmidt syndrome (polyglandular autoimmune syndrome type II. Der Hautarzt. 2001;52:974-976.
- Wollina U, Schreiber G. Polyglandular autoimmune syndrome type II (Schmidt’s syndrome) in patients with autoimmune connective tissue disorders. J Eur Acad Dermatol Venereol. 2003;17:371-372.
- Dittmar M, Kahaly GJ. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up. J Clin Endocrinol Metab. 2003;88:2983-2992.
- Wehbe E, Grant ME. Severe hyponatremia and Schmidt’s syndrome. Clin Exp Nephrol 2008;12:211-214.