Introduction
Acute primary hyperparathyroidism is a rare but potentially fatal endocrine emergency if it goes unrecognised and untreated. Prompt recognition and timely management of hypercalcemia are essential in reducing mortality with surgery being the cornerstone of definitive treatment. Primary hyperparathyroidism is often diagnosed incidentally due to raised serum calcium however it may also present with a multitude of vague and seemingly unrelated symptoms due to its effects on multiple organ systems. We present this case to emphasize the challenges of an early diagnosis, the importance of immediate management and the potential sequelae of hypercalcemia.
Case Report
An 83 year old lady was admitted with a one week history of non-specific symptoms of malaise, anorexia and epigastric discomfort along with new onset of shortness of breath for two days prior to presentation. On further questioning, she also admitted to recent symptoms of polydipsia and polyuria. She admitted to being constipated for the last number of years also. She denied weight loss, fevers, night sweats or steatorrhoea. She had not experienced chest pain, palpitations, orthopnoea or ankle swelling. Her family reported the patient had experienced low mood and some memory impairment in recent years. The patient was otherwise well, with no past medical history, except for cholelithiasis visualized on ultrasound imaging, twenty years prior. Prior to presentation, she was on no regular medications.
Family history was non-contributory. She was a lifelong non-smoker and did not drink alcohol. She had been independent of activities of daily living with excellent baseline mobility up until the two weeks preceding her admission when she had started to become dependent on a walking frame to mobilize due to global weakness. On admission her O2 saturations were 95% on room air with respiratory rate of 18 breaths per minute. Her blood pressure was 91/56 mmHg and she had developed presumably new atrial fibrillation on ECG at a rate of 105-110/min, with normal QT interval. Her ECG also showed left bundle branch block and as there was no old ECGs for comparison, she was triaged according to the acute coronary syndrome protocol and was brought immediately to the cardiac catheterization laboratory. Her angiogram revealed a 70% stenosis in her left anterior descending coronary artery. Her serial data showed troponin T of 43 ng/mL (0-14 ng/mL) rising to 70 ng/mL. As such her ECG changes were deemed by Cardiology to represent an old LBBB and she didn’t meet criteria for an acute coronary syndrome with subsequent need for immediate revascularisation. Her admission bloods however also showed an unexpected markedly elevated serum calcium of 4.16 mmol/L. She had no previous laboratory investigations available for comparison. Her laboratory investigations on admission are mentioned in Table 1 other supportive investigations are mentioned in Table 2.
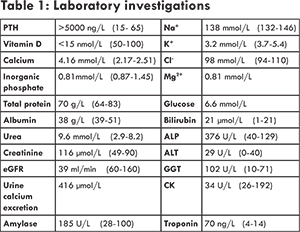

Initially the differential diagnosis included that of an acute coronary syndrome given the possible new left bundle branch on ECG and presentation with new onset shortness of breath and epigastric discomfort. Once laboratory investigations returned demonstrating the marked hypercalcaemia, the differential included parathyroid dependent hypercalcaemia versus hypercalcaemia of malignancy. The differential for parathyroid dependant hypercalcaemia was a parathyroid adenoma, gland hyperplasia or parathyroid carcinoma. Parathyroid carcinoma, whilst extremely rare, was considered a possibility in this case given the degree of hypercalcaemia, as well as the massively elevated PTH level >5000 ng/L and the newly discovered pulmonary emboli, lending further suspicion to an incidental diagnosis of malignancy. When initially faced with hyperparathyroidism along with concomitant CT imaging suggestive of a cystic pancreatic lesion, we did consider an underlying genetic syndrome such as, multiple endocrine neoplasia type 1 in our differential diagnosis. However, CT pancreatic protocol, demonstrated the pancreatic lesion to likely represent a pseudocyst from chronic pancreatic inflammation rather than a neuroendocrine tumor [Fig.2] tumor.
Initial management was directed in order of most life-threatening issues first; the possibility of a new LBBB on ECG in setting of dyspnoea and subacute upper epigastric discomfort, necessitated immediate coronary angiography. The burden of coronary artery disease seen at angiography was not felt to explain her acute presentation, and as such she did not undergo percutaenous coronary intervention acutely. The Cardiologist planned to repeat this procedure with fractional flow reserve measurement (FFR) at a later date if her clinical symptoms persisted.
However once the impressive hypercalcaemia was discovered on routine biochemistry, attention turned to the acute management of a calcium level of 4.16 mmol/L (2.17-2.51 mmol/L). Intravenous (IV) fluids were commenced immediately. Following this, once she was intravascularly volume replete, IV furosemide (loop diuretic) was administered in order to increase urinary calcium excretion. Given the degree of hypercalcaemia, she was then also given a bisphosphonate, IV zoledronic acid 4 mg. Within 2-3 days her serum calcium started to respond and had decreased to 2.7 mmol/L after 72 hours of initial presentation.
The next acute issue was the subsequent development of hypomagnesemia felt to be secondary to the excess use of IV normal saline causing distal tubule dysfunction with urinary magnesium loss. We exercised caution when replacing her magnesium cognisant of the biochemical relationship between magnesium, calcium and PTH. Under normal circumstances, increasing magnesium will increase PTH release which subsequently will further increase calcium release from bone and aggravate hypercalcaemia. In our case, the PTH secretion was felt to be under autonomous control, and so the influence of magnesium causing a further increase in PTH would be a lot less, however we were still mindful of the above when replacing her magnesium intravenously, aiming to keep the magnesium level at the lower limit of normal necessitating frequent monitoring of her serum calcium and magnesium levels. In addition, given the need for aggressive use of IV diuretics and normal saline in order to treat the life-threatening hypercalcaemia, the patient’s serum potassium subsequently dropped to 2.8 mmol/L (3.5-5.1 mmol/L), also requiring IV replacement. Such significant electrolyte disturbances are a major contributor to the development of cardiac arrhythmias and as such continuous cardiac monitoring using telemetry was a priority for our patient.
The patient’s CT aortic angiogram had revealed significant and unexpected pathology, including pulmonary emboli, nephrocalcinosis and a pancreatic lesion. Dedicated abdominal CT imaging fortunately ruled out a pancreatic cystic neoplasm and instead demonstrated the lesion to represent a simple pseudocyst attributed to a previous episode of pancreatitis. A sestamibi isotope parathyroid scan using technetium-99m revealed markedly increased radioisotope uptake by the hypoechoic nodule, previously visualized on ultrasound, in the left neck posterior to the left lobe of thyroid gland, felt to be consistent with a parathyroid adenoma.
Our patient underwent an uneventful minimally invasive left lower parathyroidectomy two weeks after her initial presentation. Given the significantly elevated PTH level of >5000 ng/L, the calcium of 4.16 mmol/L and the finding of pulmonary emboli, we were initially suspicious that this may represent the rare entity of a parathyroid carcinoma and it was of utmost importance that our surgical colleagues were aware of this possibility prior to resection. However, the surgical team reported that macroscopically the parathyroid gland did not appear malignant and was excised with clear margins from surrounding structures. Post-operatively our patient was monitored in a high dependency setting, continued on cardiac monitoring, with frequent measurements of her serum and ionised calcium levels. Her postoperative initial serum calcium was 2.12 mmol/L (2.17- 2.51 mmol/L). We observed closely for development of any symptoms of hypocalcemia such as paraesthesia, muscle cramps, twitching or carpopedal spasm. IV magnesium was also promptly replaced postoperatively.
Day 1 postoperatively the patient’s serum calcium dropped to 1.95 mmol/L and as such we treated her according to our hospitals’ protocol for management of significant post-parathyroidectomy hypocalcemia. We administered an initial bolus of IV 2.2 mmol of calcium gluconate followed by an IV infusion of (1.7 x patient’s weight in kg) mls of 10% calcium gluconate. In our patient’s case, we required 10 vials of 10 mls of calcium gluconate; 22 mmols, in 1 litre of normal saline to be given over eight hours. We also commenced Alfacalcidiol, an activated form of vitamin D was also initiated in the setting of her renal impairment, to try to increase her calcium absorption. Treatment of her incidentally diagnosed pulmonary emboli was initially with therapeutic low molecular weight heparin, pending a date for surgery.
Following successful parathryoidectomy and stabilisation of her serum calcium, the patient was fit for discharge home one week postoperatively. Discharge prescription included alfacalcidiol (one alpha hydroxy-vitamin D3) 0.5 mcg twice daily and three calcium carbonate tablets (1500 mg) three times daily. We planned to regularaly monitor electrolytes twice weekly initially and more frequently if dictated by development of symptoms, and then once weekly until calcium, magnesium and Vitamin D levels remained stable.
The length of follow-up at this time has been one month. The patient is doing well, with slight adjustments having been made to her medication regimen and continues now to be closely followed in our Endocrinology outpatient department for monitoring of her electrolyte levels. Her parathyroid histology has returned and is consistent with a benign parathyroid adenoma, and thus she should now have a surgical cure from her primary hyperparathyroidism. She will continue warfarin therapy for six months and will need close follow up with her primary doctor to monitor her INR (international normalised ratio). Given her hyperparathyroidism of unknown duration, she is at an above average risk for low bone mineral density and subsequent osteoporosis. She has not had any reported fractures in the past and has never had a DEXA scan performed. She did have an X-ray of her shoulder performed during her inpatient stay after complaining of shoulder pain but there was no fracture identified. Of note her CT scan did reveal age indeterminate fractures of the right superior inferior pubic rami. A number of other tests of diagnostic interest were also considered including an isotope bone scan and plain radiographs of her thoracolumbar spine to assess for silent vertebral fractures, however the patient declined further extra investigations during her inpatient stay due to desire to return home. She is now awaiting a DEXA scan and isotope bone scan for further evaluation, but would like first to recover from her hospitalization.
In terms of the radiologically confirmed pancreatitis and pancreatic pseudocyst, her epigastric pain has fully resolved and serum amylase is with normal range. We would anticipate that she should have no further episodes of pancreatitis now that the inciting stimulus of hypercalcaemia has been removed. At this present time, she has no symptoms of pancreatic insufficiency but healthcare professional should be aware to carefully screen for features of steatorrhoea and malabsorption during future follow-up.
Discussion
This case highlights the diagnostic difficulty when a patient, never having had previous medical investigation, presents with vague, non-specific symptoms and thus old laboratory results or ECGs are not available to serve as a reference point. This interesting case of a patient with primary hyperparathyroidism who had likely been hypercalcemic for a significant period of time prior to presentation can educate us on some of hypercalcaemia’s protean complications that include nephrolithiasis, osteopenia, pancreatitis and tooth resorption [1].
Management of symptomatic primary hyperparathyroidism is relatively straightforward; surgical parathyroidectomy by a surgeon experienced in this area representing the definitive treatment. For those with asymptomatic primary hyperparathyroidism management guidelines are currently based on the Fourth International Workshop on Asymptomatic Primary Hyperparathyroidism guidelines, published in the Journal of Clinical Endocrinology Metabolism in 2014 [2], with surgery advised in i) patients with a serum calcium level > 0.25 mmol/L above normal level, ii) patients less than 50 years of age, iii) patients with an eGFR < 60 ml/min, iv) patients with a T score < -2.5 and/or previous asymptomatic vertebral fracture, v) patients with nephrolithiasis or nephrocalcinosis on imaging and vi) patients with urinary calcium excretion >10 mmol/day.
Primary hyperparathyroidism is considered by some to be an almost benign entity, as there exists a relatively routine surgical cure. However this case reminds us of the numerous complications that may arise in the setting of hyperparathyroidism and its consequent hypercalcaemia. The clinical syndrome of primary hyperparathyroidism is often taught to medical students using the mnemonic first described by Jackson and Frame in 1972 as “Bones, stones, abdominal groans, and psychic moans with fatigue overtones”. Skeletal manifestations include selective cortical bone loss, which is believed to be the most common. According to the literature radiographic skeletal surveys are normal in most cases of primary hyperparathyroidism, but specific changes such as subperiosteal erosions may occasionally be seen [3]. According to an article published in Nuclear Medicine Review in 2012 bone scans usually show slight generalised increased uptake with high bone to soft tissue ratios. The degree of bone scan abnormality generally reflecting the amount of skeletal involvement and there is thus a wide range of scan appearances from normal to those mimicking severe renal osteodystrophy [3]. Bone and joint pain, pseudogout, and chondrocalcinosis have also been reported. In the early clinical description of primary hyperparathyroidism, some patients developed a peculiar type of bone disease known as Von Recklinghausen disease or osteitis fibrosa cystica, characterized by increased generalized osteoclastic bone resorption, particularly involving the phalanges causing subperiosteal resorption, and the skull causing the ‘salt and pepper’ appearance on X-ray [4]. Oral manifestations include but are not limited to external and internal root resorption of teeth and jaw lesions such as cysts, fibro-osseous lesions and odontogenic tumours [1,5]. This is much less common in the current era, given the earlier recognition of primary hyperparathyroidism and subsequent hypercalcemia on biochemical testing.
Renal manifestations include polyuria, hypercalciuria, nephrolithiasis and indeed nephrocalcinosis, which had developed in our patient’s case. Gastrointestinal manifestations include anorexia, nausea, vomiting, vague abdominal pain, constipation, peptic ulcer disease, and acute pancreatitis. The incidence of acute pancreatitis in patients with primary hyperparathyroidism is reported to be 1.5% [6]. In 2012 in the Journal of Clinical Gastroenterology [7], a literature review was performed on the association of primary hyperparathyroidism with pancreatitis, which identified 10 retrospective studies each with >50 patients diagnosed with primary hyperparathyroidism (PHPT), and with the exception of two studies, the rate of pancreatitis among patients with PHPT was higher than that reported overall among hospitalized patients without PHPT.
Central nervous system and neuromuscular symptoms are less easy to recognize. These include tiredness, headache, components of an endocrine psychologic syndrome (e.g., loss of initiative), depression, inability to concentrate, memory problems, weakness and proximal myopathy. Cardiovascular manifestations include hypertension, bradycardia, shortened QT interval, and left ventricular hypertrophy. Our patient was found to be hypertensive which had been untreated prior to this presentation and also had ECG changes of LBBB which can be associated with hypertensive heart disease.
This case highlights the importance of recognizing that this degree of hypercalcemia is a medical emergency requiring immediate treatment and very close patient monitoring. When hypercalcemia reaches a critical level (>4 mM), two organs are at risk for decompensation; the kidneys and the brain, termed a hypercalcemic crisis [8]. Decompensated primary hyperparathyroidism is felt to be the commonest entity causing a hypercalcemic crisis. Polyuria may develop into oliguria and finally anuria, especially in the case of exsiccosis. If not treated promptly, hypercalcemic renal insufficiency can be fatal. Psychologic disturbances may develop into somnolence and finally coma. Approximately 20 years ago, there were more reports of hypercalcemic crisis [9]. It is evident that the current use of early intravenous fluids for critically ill patients and optimum strategies for intensive care medicine have made hypercalcemic crisis now a rare event. As is now the standard of care, our patient’s hypercalcemia was treated efficiently with IV fluids, IV loop diuretics once she had been rendered intravascularly volume replete followed by IV bisphosphonate therapy.
Fortunately our patient did not run into complications of a life-threatening hypercalcemic crisis, however post-operatively we had to be aware of the potential for development of hungry bones syndrome. The “hungry bone disease” occurs in consequence of the rapid accrual of calcium from the intravascular compartment into the skeleton and under these conditions can result in severe hypocalcemia and the urgent need for intravenous calcium replacement [10]. An update on the hungry bone syndrome published in 2007 in Germany [11], found that risk factors for its development included; large parathyroid adenomas, age >60 years, high preoperative levels of serum PTH, calcium and alkaline phosphatase. A paper published in the American Journal of Surgery in 2004 looked at 162 cases of parathyroidectomy and found 52% were hypocalcemic postoperatively [12]. They found patients with renal hyperparathyroidism were more profoundly hypocalcemic, as were patients who underwent a subtotal parathyroidectomy rather than single or double adenoma removal. Bisphosphonates, which we administered to our patient prior to surgery as part of management of her hypercalcaemia, also help to decrease bone turnover (bone resorption and bone formation) and to prevent, therefore, the development postoperatively of severe hypocalcemia and its complications such as seizures and coma.
Ensuring calcium and vitamin D supplementation both postoperatively and over subsequent months is essential in this situation. It may be interesting to monitor urinary calcium levels in her future care in monitoring her dosing of oral calcium, especially given the fact she had evidence of nephrocalcinosis at presentation. Her vitamin D level had not returned prior to surgery, but had been low; below the recommended 50 nmol/L. Interestingly the association between low vitamin D levels and primary hyperparathyroidism is being increasingly recognized. Recent publication in the Journal of Endocrinological Investigation [13], describes findings of specific patient populations with primary hyperparathyroidism and concomitant vitamin D deficiency who have been found to have higher PTH levels, larger adenomas and a more severe phenotype with increased bone turnover and low bone mineral density measurements. This population of patients were also reported to have a trend towards more negative post-operative outcomes following parathyroidectomy. Further research is needed to determine if repletion of vitamin D, whether pre or post-operatively, could improve such patient’s overall clinical outcomes.
Our patient’s advanced age was also considered when deciding on a management plan. Literature on hyperparathyroidism in elderly populations is limited, comparative to that of middle-aged cohorts. However the incidence of primary hyperparathyroidism does in fact increase with age. The treatment decision on whether to proceed to surgery was relatively straightforward in our patient’s case; she had relatively few co-morbidities , her hyperparathyroidism had resulted in a calcium level that had the potential to be life-threatening if not treated and parathyroidectomy represented a definitive surgical cure. A recent publication [14] supports such a decision, reporting that with the exception of a few cases with severe associated comorbidities, parathyroidectomy is safe and curative in an elderly population (aged over 75), and should also be considered as first-line choice for older adults with PHPT.
Another discussion point in this case is that parathyroid carcinoma was considered in the differential diagnosis for this patient based on the excessively high PTH level >5000 nmol/L and serum calcium level above 4 mmol/L as well as the finding of pulmonary emboli. Parathyroid carcinoma accounts for less than 1% of cases of primary hyperparathyroidism but if suspected needs to be highlighted pre-operatively to the surgical team. Conventional neck exploration is indicated in patients suspected of having a parathyroid carcinoma. Intraoperative findings are important to distinguish between an adenoma and a carcinoma. A parathyroid carcinoma generally has a fibrous capsule and a grayish colour, and is often tenaciously adherent to the adjacent tissues eg. thyroid, recurrent laryngeal nerve and overlying muscle [15]. Parathyroid carcinomas are usually located in the inferior parathyroid glands. When there is no preoperative sign of parathyroid carcinoma, minimally invasive adenectomy, as in our case, is the procedure of choice. If possible, only the involved parathyroid gland should be removed, without rupturing the capsule, but if during surgery, invasive growth is suspected, en bloc resection of the ipsilateral thyroid gland is essential, as is the segmental removal of the overlying muscles of the paratracheal fibrolymphatic compartments and the remaining tissue around the involved parathyroid gland [16].
Conclusion
Hypercalcemia is an medical emergency that should be considered in the differential for non-specific symptom presentation and promptly treated. Development of ‘hungry bones’ post-operatively in a patient with chronically elevated calcium needs close electrolyte monitoring in the post-operative setting.
References
- Nagaraj E, Kaur RP, Raghuram PH, Kumar PS. Multiple internal resorption in permanent teeth associated with hyperparathyroidism. Indian J Dent Res. 2013;24(1):128-131.
- Bilezikian. JP, Brandi. M, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, et al. Guidelines for the Management of Asymptomatic Primary Hyperparathyroidism: Summary Statement from the Fourth International Workshop. The Journal of Clinical Endocrinology and Metabolism. 2014; 99(10):3561-3569.
- Abdelrazek S, Szumowski P, Rogowski F, Kociura-Sawicka A, Mojsak M, Szorc M. Bone scan in metabolic bone diseases. Review. Nuclear Medicine Review. 2012;152:124-131.
- Kim L. Hyperparathyroidism. e-medicine. Medscape. Available at: http://emedicine.medscape.com/article/127351-overview. Accessed August 30, 2015.
- Shaveta G, Ashwarya T, ParamPal S, Neetu S, Beant K. Osteodystophic lesions of the jaw-a review. Journal of PEARLDENT. 2014;5(2):14-24.
- Carnaille B, Oudar C, Pattou F, Combemale F, Rocha J, Proye C. Pancreatitis and Primary hyperparathyroidism: Forty cases. Aust N Z J Surg. 1998;68:117-119.
- Bai HX, Giefer M, Patel M, Orabi AI, Husain SZ. The association of primary hyperparathyroidism with pancreatitis. J Clin Gastroenterol. 2012;46:656-661.
- Ziegler R. Hypercalcemic crisis. J Am Soc Nephrol 2001;12(Suppl 17):S3-S9.
- Binstock ML, Mundy GR. Effect of calcitonin and glucocorticoids in combination on the hypercalcaemia of malignancy. Ann Intern Med. 1980;93(2):269-272.
- Bandeira F, Cusano N , Silva BC, Cassibba S, Almeida CB, Machado VCC, et al. Bone disease in primary hyperparathyroidism Arq Bras Endocrinol Metab online, 2014;58(5):553-561.
- Farese S. The hungry bone syndrome - an update. Ther Umsch. 2007;64:277-280.
- Mittendorf EA, Merlino JI, McHenry CR. Postparathyroidectomy hypocalcemia: incidence, risk factors, and management. Am J Surg. 2004;70:114-119.
- Nuti R, Merlotti D, Gennari L. Vitamin D deficiency and primary hyperparathyroidism. J Endocrinol Invest. 2011;34(7 Suppl):45-49.
- Denizot A, Grino M, Oliver C Surgical Management of Primary Hyperparathyroidism in Older Adults. Journal of the American Geriatrics Society. 2014;62 (9):1759-1763.
- Mittendorf EA, McHenry CR. Parathyroid carcinoma. J Surg Oncol. 2005;89(3):136-142.
- Ter Haar RW, Shen WT, Valk GD, Borel Rinkes IHM, Vriens MR. Parathyroid Carcinoma, a Rare Entity with Varying Presentation and Treatment. World Journal of Endocrine Surgery. 2010;2(1):33-36.