Introduction
Arthrogryposis-Renal dysfunction-Cholestasis (ARC) syndrome is a lethal, autosomal recessive, multi-systemic disorder [
1]. It not so rare syndrome rather underdiagnosed due to wide phenotypic variability of the syndrome [
2,
3]. Our case apart from cardinal features also had seborrheic dermatitis, scalp hypotrichosis and choroidal sclerosis as associated features. The case also highlights that in view of fatal outcome of disease early detection with supportive treatment and genetic counselling is also important.
Case Report
Two month old female child born of non-consanguineous marriage, second by birth order presented with history of fever for 4 days, jaundice since 8th day of life, failure to thrive, dryness of skin and restricted movements of lower limbs since birth. On examination anterior fontanelle 1.5x1.5 cm, seborrheic dermatitis, scalp hypotrichosis, icterus, choroidal sclerosis, low set ears, micrognathia, radial deviation of wrist, rocker-bottom feet, overlapping of fourth toe, exfoliative skin, ichthyosis, sacral sinus were seen [Fig.1-4]. Anthropometry showed weight: 3.2 kg (< 3rd centile) and length: 51 cm (< 3rd centile). No history of any dysmorphic features or jaundice in sibling or family could be elicited. Hepatomegaly with span 9 cm was noted and rest of systemic examination was normal. Routine haematological and biochemical investigations were hemoglobin 6.6 g/dL, white blood cells: 13800/cu.mm, platelet count: 380,000/cu.mm, blood urea nitrogen: 12 mg/dL and serum creatinine 0.6 mg/dL, serum bilirubin 6.7 mg/dL with direct bilirubin 3.3 mg/dL, alanine transaminase: 28 IU/L and aspartate transaminase: 24 IU/L, gamma glutamyl transaminase: 8 IU/L (7-98 IU/L). Acid-venous blood gas analysis was suggestive of normal anion gap metabolic acidosis, urine reducing substances were positive, radionuclide scan showed excretion in intestine. Peripheral smear showed giant platelets [Fig.5]. Histopathology of liver showed paucity of bile ductules [Fig.6]. Ultrasound of sacral region suggested a blind sinus. 2D Echo, brainstem enhanced response audiometry and MRI brain of child was normal. The child was treated with ursodeoxycholic acid, fat soluble vitamins and liquid paraffin for local application on skin. The child initially improved with respect to decrease in icterus, however, succumbed to severe pneumonia at the age 6 months.
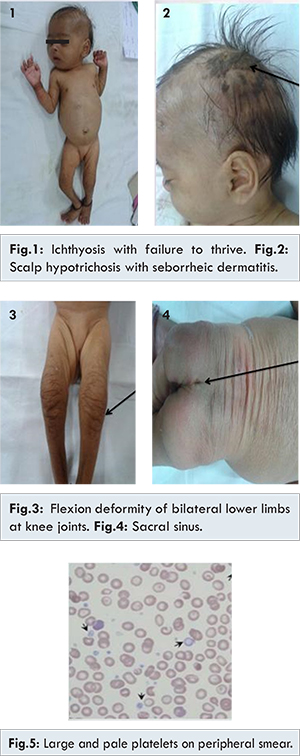
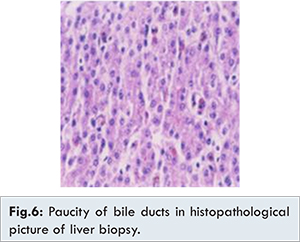
In this case the child presented with restricted movements of lower limbs, rocker bottom feet, dysmorphic facies, cholestasis, with paucity of bile duct, ichthyosis, and renal dysfunction in form of isolated metabolic acidosis, failure to thrive, morphological abnormality of platelets in form of large and pale platelets, suggestive of ARC syndrome. Additional features in our case were choroidal sclerosis, scalp hypotrichosis and seborrheic dermatitis.
Neonatal ichthyosis-sclerosing cholangitis (NISCH syndrome) also has ichthyosis, scalp hypotrichosis, fever and bile duct paucity but clinching the diagnosis was demonstration of large platelets on peripheral smear [
4]. Congenital ichthyosis and hepatomegaly also made us consider Chanarin Dorfman syndrome but absence of lipid filled vacuoles in leucocytes ruled it out [
5].
ARC syndrome is caused due to germline mutation in gene at locus 5q26.1 known as vacuolar protein sorting 33 homolog B (VPS33B) in 75% of cases and mutation in VPS33B- interacting, apical basolateral polarity regulator (VIPAR). VPS33B protein is a member of SNARE (sensory nerve action potential receptor) family involved in vesicular trafficking and membrane fusion. The mutated VPS33B-VIPAR gene product fails to conserve the apical basolateral polarity so the tissue structure is affected like that in biliary ducts hampering transport of bile and renal involving both podocytes and glomerular endothelial cells leading to cholestasis and abnormal urine also in abnormalities in other systems [
2,
6,
7].
Arthrogryposis though a principal symptom has a wide range of manifestations like neurogenic muscle atrophy, lateral deviation of wrist, dislocation of bilateral hip joints and talipes equanovarus deformity, all these changes are primarily due to anterior motor neuron cell degeneration [
2]. Our case had rocker bottom feet and only mild flexion deformity of knee joints. Renal tubular dysfunction spectrum ranges from Fanconi syndrome, nephrogenic diabetes insipidus, aminoaciduria, phosphaturia, and nephrocalcinosis [
2]. Our child had isolated renal tubular acidosis which is commonly seen. Neonatal cholestasis is characteristically associated with low GGT as seen in our case [
6]. Liver biopsy done in our case had no bleeding complications, seen in about 50% of cases with liver biopsy. Other histological features seen include giant cell transformation, deposition of lipofuscin, bile plugs, fibrosis and cirrhosis [
2].
Additional features like ichthyosis in half of patients is faulty differentiation of epidermis because of lack of absorption of free fatty acids [
8]. Large, pale and agranular platelets are seen in about a fourth of ARC syndrome patients, can be associated with absent or abnormal alpha granules on electron microscopy [
9]. Other features are agenesis of corpus callosum (20%), congenital heart diseases (10%), sensory neural deafness, spontaneous life threatening bleeding and hypothyroidism [
2,
10]. Recurrent infections are seen due to phagosome lysosome defect [2].
Diagnosis in our case was made with clinical picture, liver biopsy findings, and platelet abnormalities. Mutational analysis can be done but has drawbacks like having high false negatives and takes long duration for results. Hence the expression VPS33B protein in skin fibroblasts can be studied [
2,
11]. Specific treatment for this disorder does not exists, so supportive care apart from ursodeoxycholic acid and fat soluble vitamins, medium chain triglycerides, L-thyroxin, phosphate and calcium can be given [
2]. Liver transplantation can be tried in cases of severe cholestasis and troublesome itching [
12].
The prognosis of this disorder is extremely poor, as patients die in infancy. Most deaths are due to recurrent infection, acidosis and critical bleeding [
2]. Hence we conclude that there is large spectrum of clinical presentation, early identification of this disorder aid to customise treatment, no major response to current treatment strategies, counselling of the parents regarding the present child as well as pre-natal and pre-implantation is of immensely vital.
References
- Li L, Zhao J, Chen R, Wang J. Two novel VPS33B mutations in a patient with arthrogryposis, renal dysfunction and cholestasis syndrome in mainland China. World J Gastroenterol. 2014;20(1):326-329.
- Zhou Y, Zhang J. Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: from molecular genetics to clinical features. Ital J Pediatr. 2014;40:77.
- Malaki M, Mandana R, Ghaffari S. ARC syndrome with complex renal problems: nephrocalcinosis, proximal and hyperkalemic distal RTA and nephrogenic diabetes insipidus. Saudi J Kidney Dis Transpl. 2012;23(4):804-809.
- Nagtzaam IF, Van Geel M, Driessen A, Steijlen PM, Van Steensel MA. Bile duct paucity is part of the neonatal ichthyosis-sclerosing cholangitis phenotype. Br J Dermatol. 2010;163(1):205-207.
- Ben Ameur S, Aloulou H, Wali M, Alibi S, Chaari M, Kallel C, et al. Congenital ichtyosis and hepatomegaly: Think about Chanarin Dorfman. Tunis Med. 2015;93(7):2015.
- Wang JS, Zhao J, Li LT. ARC syndrome with high GGT cholestasis caused by VPS33B mutations. World J Gastroenterol. 2014;20(16):4830-4834.
- Holme A, Hurcombe JA, Straatman-iwanowska A, Inward CI, Gissen P, Coward RJ. Glomerular involvement in the arthrogryposis, renal dysfunction and cholestasis syndrome. Clin Kidney J. 2013;6(2):183-188.
- Elmeery A, Lanka K, Cummings J. ARC syndrome in preterm baby. J Perinatol. 2013;33(10):821-822.
- Saadah OI, Bokhari BE, Alshaeri TM, Jastaniah W. Haematological manifestations of arthrogryposis-renal dysfunction- cholestasis (ARC) syndrome: a case report. Arab J Gastroenterol. 2013;14(1):26-28.
- Kim SM1, Chang HK, Song JW, Koh H, Han SJ. Severance Pediatric Liver Disease Research Group. Agranular platelets as a cardinal feature of ARC sysdrome. J Pediatr Hematol Oncol. 2010;32(4):253-258.
- Dehghani SM, Bahador A, Nikeghbalian S, Salahi H, Geramizadeh B, Malekpour A, Malek-Hosseini SA. Liver Transplant in a case of Arthrogryposis-renal tubular dysfunction-cholestasis syndrome with severe intractable pruritus. Exp Clin Transplant. 2013;11(3):290-292.