Introduction
Hemimegelencephaly (HME) or unilateral megelencephaly is rather an unclassified malformation of cortical development (MCD) regarded as a result of abnormal neuronal migration that occurs during early gestation between 3rd and 5th week of life [
1-
3]. Isolated form is the most common type, which usually presents during first month of life with head or facial asymmetry. We present an isolated case of a full term phenotypically normal neonate with generalized tonic clonic seizure on day three. MRI of brain showed hypertrophy of right cerebral hemisphere with dilatation of lateral ventricle with normal contralateral cerebral hemisphere which clinched the diagnosis of right hemimegelencephaly. He was started on anti-epileptics and there was no further episode of seizure during hospitalization and follow up.
Case Report
Our case is a three days old male neonate, first born, out of a full term normal vaginal delivery who presented to emergency with first episode of generalized tonic-clonic seizure at 72 hours of life. Antenatal history and perinatal history was uneventful. Parental history was also negative for neurological illness. Baby had been on exclusive breastfeeds since birth. Clinically he was stable with no facial dysmorphism or asymmetry of the head and body (occipito-frontal circumference was 36 cm). All fontanelles and sutures were normal. There were no neurocutaneous markers. Baby’s birth weight was 3.5 kg and length was 50 cm. Differential diagnosis considered were late onset meningitis, dyselectrolytemia, inborn errors of metabolism and central nervous system abnormalities.
Blood routine investigations including sepsis panel and metabolic workup were normal. Neuroimaging was planned to rule out intracranial pathology. Computed tomography (CT) head was done which showed unexpected finding of atrophic left hemisphere. Later, magnetic resonance imaging (MRI) was done to confirm the findings. It showed hypertrophy of the right cerebral hemisphere with dilatation of the right lateral ventricle. The right cerebellar hemisphere and brain stem was normal. The left cerebral parenchyma appeared normal, with no evidence of microvascular changes, infarction or hemorrhage [Fig.1]. Electroencephalogram (EEG) showed activation of frequent epileptiform abnormalities with high amplitude spikes arising from right hemispheric regions [Fig.2].
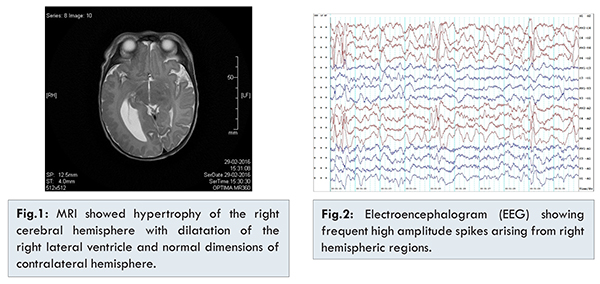
Baby was managed in neonatal intensive care unit with supportive care including intravenous fluids, empirical antibiotics and antiepileptic drugs as per the unit protocol. Baby was seizure free after hospitalization. Parental counseling was done and baby was discharged on oral levetiracetam. Baby is under regular follow up.
Discussion
Hemimegelencephaly (HME) or unilateral megelencephaly is a rare, congenital, sporadic malformation of brain in which one-half of the brain shows hamartomous overgrowth compared to the other half. It is rather an unclassified malformation of cortical development (MCD) as traditional neuroembryology is unable to explain extreme asymmetry not corresponding to any normal stage of human brain development - neuronal proliferation, migration and organization [
1,
2]. However, it is regarded as a result of abnormal neuronal migration that occurs during early gestation between 3rd and 5th week of life [
3]. It was first described by Sims in 1835 as a malformation of cerebral cortical development based on the autopsy record of 235 cases [
4]. In past decades, there has been interest in genetic studies involving genes responsible for right-left symmetry of the brain neuraxis. A recent report has shown de novo somatic mutations in components of the PI3K-AKT3-mTOR pathway as a cause of hemimegelencephaly [
5].
Three types of hemimegelencephaly have been described [
6]. Isolated form is the most common and occurs as a sporadic disorder without skin or systemic involvement. Our case report was consistent with this form. Syndromic hemimegalencephaly [
7] is associated with other neurocutaneous syndrome like epidermal nevus syndrome, Proteus syndrome, encephalocraniocutaneous lipomatosis, hypomelanosis of Ito, Klippel-Trenaunay syndrome and rarely tuberous sclerosis [8]. Least common form is total hemimegelencephaly where there is also enlargement of the ipsilateral half of the brain stem and cerebellum [
9].
Clinical picture of hemimegelencephaly varies. Available literature does not show any predisposition of ethnic groups and no male and female difference. Neurological presentation is usually moderate to severe in all forms and correlate with involvement of affected cerebral hemisphere. During first month, presentations have been macrocephaly, facial or head asymmetry and seizures. Syndromic forms and some isolated forms present during first month with severe phenotypes and are associated with progressive neurological damage. Global developmental delay is noted. Seizures refractory to treatment may require surgical hemispherectomy during first few months of life. Further brain development and prognosis depends on the functional status of contralateral cerebral hemisphere [
6].
Our case is consistent with mild isolated hemimegelencephaly with no asymmetry of head, facial dysmorphism or neurocutaneous markers. Prognosis of such cases varies. Some have normal intellectual development but may have pathological left-handedness, febrile seizures, learning disabilities, and clumsiness. Lifelong antiepiletics are needed in those having partial epilepsy. Parental counselling is important in cases presenting early neonatal period, especially those with mild grades of severity. There are reports of rapid, progressive hydrocephalus in first few months [
10]. An unusual arrested growth or even atrophy of the affected hemisphere can occur in due course [
11].
Conclusion
Isolated hemimegelencephaly of mild severity presenting during neonatal period is challenge to the clinician. Most of these cases follow an asymptomatic course and prognosis depends on functional status of contralateral cerebral hemisphere. However, there are certain areas of concern especially parental counselling and need for regular follow up.
Acknowledgments
We would also like to show our gratitude to our neurologist Dr. Dinesh Kamath for sharing his pearls of wisdom with us during the course of this manuscript preparation, and our radiologist Dr. Asghar Majeed for his valuable inputs.
References
- Sarnat HB. Cerebral Dysgenesis. New York: Oxford University Press; 1992.
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development. Update Brain. 2012;135:1348-1369.
- Battaglia D, DiRocco C, Iuvone L, Acquafondata C, Lanelli A, Lettori D, et al. Neuro-cognitive development and epilepsy outcome in children with surgically treated hemimegalencephaly. Neuropediatrics. 1999;30:307-313.
- Sims J. On hypertrophy and atrophy of the brain. Med Quir Trans. 1835;19:315-380.
- Lee JH, Huynh M, Silhavy JL, Kim S, Salazar TD, Heiberg. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nature Genetics. 2012;44:941-945.
- Laura Flores-Sarnat. Hemimegalencephaly: Part 1. Genetic, Clinical, and Imaging Aspects. Child Neurol. 2002;17:373-384.
- Tinkle BT, Schorry EK, Franz DN, Crone KR, Saal HM. Epidemiology of hemimegalencephaly: a case series and review. Am J Med Genet A. 2005;139:204-211.
- Sakuma H, Iwata O, Sasaki M. Longitudinal MR findings in a patient with hemimegalencephaly associated with tuberous sclerosis. Brain & Development. 2005;27:458-461.
- Hallervorden J. Angeborene Hemihypertrophie der linken Körper hälfte inschliesslich des Gehrins. Zentralbl Neurol Psychiatr. 1923;33:518-519.
- Broumandi DD, Haywa UM, Benzian JM, Gonzalez I, Nelson MD. Best cases from the AFIP: hemimegalencephaly. Radiographics. 2004;24:843-848.
- Wolpert SM, Cohen A, Libenson MH. Hemimegalencephaly: A Longitudinal MR Study. Am J Neuroradiol. 1994;15:1479 -1482.