6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff70fe1e0000003c07000001000800
6go6ckt5b5idvals|820
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Noncompaction cardiomyopathy or "spongy myocardium", is a rare congenital cardiomyopathy that can be diagnosed at any age. It is characterized by a thin, compacted epicardial layer and an extensive non-compacted endocardial layer, with prominent trabeculation and deep recesses that communicate with the left ventricular cavity but not with the coronary circulation [
1], probably due to an arrest of compaction during intrauterine life. It can be isolated or associated with other congenital diseases. Based on echocardiographic studies, reported prevalence is between 0.014 and 1.3% in the general population. Eventually, this condition can potentially lead to chronic heart failure, life threatening ventricular arrhythmias and systemic embolic events [
2].
Case Report
A 13-year-old boy presented with a two day history of abdominal pain after being hit by his friend. He was previously healthy, referring no prior cardiac or pulmonary complaints. His father died of gastric cancer at the age of 40, his elder brother died of heart failure when he was 13 years old. He was referred from a hospital with provisional diagnosis of dilated cardiomyopathy and treated for heart failure. On admission to the Emergency Ward of our hospital, his vital signs were blood pressure: 80/48 mmHg, pulse: 78 bpm, SpO2 97% (air room), temperature 37ºC. Patient clinically stabilized within next 24 hours and was referred to the Cardiology Ward. Physical examination showed point of maximum impulse at the sixth intercostal space and anterior axillary line, S3 and a grade III/VI holosystolic murmurs in apical; liver edge below 2 cm the right costal margin.
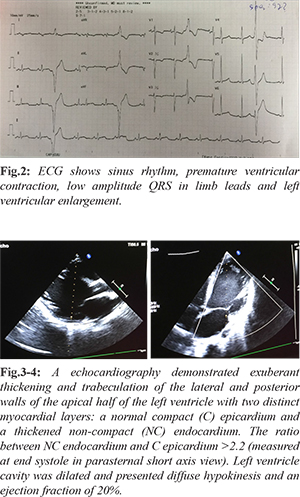
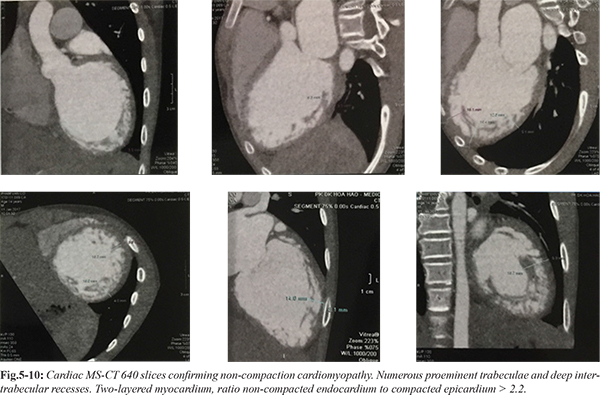
Chest X-ray demonstrated increased heart-thorax index [Fig.1]. ECG show right axis deviation, enlargement of both atria, hypertropihy of both ventricles, and ventricular premature contraction [Fig.2]. Holter ECG showed sinus tachycardia, isolated ventricular premature contraction was 17% (25,219), 4 beats fastest run 227 bpm. TSH was 1.84 µIU/mL (normal value 0.35-4.94 µIU/mL). HDL-C was low 0.75 mmol/L. Liver transaminases levels and renal function tests were normal. Carotid and aortic artery Doppler ultrasound were normal. An echocardiography demonstrated exuberant thickening and trabeculation of the lateral and posterior walls of the apical half of the left ventricle with two distinct myocardial layers: a normal compact (C) epicardium and a thickened non-compact (NC) endocardium. The ratio between NC endocardium and C epicardium >2.2 (measured at end systole in parasternal short axis view). Left ventricle cavity was dilated and presented diffuse hypokinesis and an ejection fraction of 20%. No additional abnormalities were found [Fig.3-4]. These findings were consistent with the diagnosis of non-compaction cardiomyopathy. A cardiac MS-CT 640 confirmed the diagnosis, revealing numerous proeminent trabeculae and deep intertrabecular recesses and a ratio between NC endocardium and C epicardium >2,2 [Fig 5-10].
Discussion
Non-compaction cardiomyopathy is considered a rare disease, although its exact prevalence is unknown. According to one of the largest series published, its prevalence was 0.014% in a group of patients referred to an echocardiography laboratory for abnormal findings or congestive heart failure [
5]. Men appear to be more frequently affected, accounting for 56 to 82% of reported cases [
5,
6]. Several diagnostic criteria have been proposed in the literature [
3]. In this clinical case diagnosis was primarly established by echocardiography according to the criteria defined by Jenni and colleagues. According to these authors diagnosis is based on the detection of two myocardial layers, with a normal compact (C) epicardium and a thickened non-compact (NC) endocardium. They propose a quantitative evaluation by determining the ratio between the 2 layers NC/C = 2, measured at end systole in parasternal short axis view, to establishe diagnosis. This allows differentiation of the trabeculations of non-compaction cardiomyopathy from that observed with dilated cardiomyopathy and hypertropic cardiomyopathy.
Conclusion
Non-compaction cardiomyopathy is a rare and recently described congenital cardiomyopathy. Its impact on morbidity and mortality is gaining importance as it is recognized with increasing frequency. An early, reliable diagnosis is crucial and echocardiography represents a prominent role. Treatment is directed towards its most important clinical implications, which include heart failure, arrhythmias and systemic embolic events. Heart transplantation is the only definitive treatment. Attending to the risk of cardiac sudden death, implantable cardioverter-defibrillator (ICD) may have a role in these patients.
Contributors: HTP wrote the manuscript, did literature search, involved in patient management and approved the final version of this article. He will act as guarantor of the study.
Funding: None; Competing interests: None stated.
References
- Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82:507-513.
- Engberding R, Bender F. Identification of a rare congenital anomaly of the myocardium by two-dimensional echocardiography: persistence of isolated myocardial sinusoids. Am J Cardiol. 1984;53:1733-1734.
- Jenni R, Oechslin EN, Loo van der B. Isolated ventricular non-compaction of the myocardium in adults. Heart. 2001;93:11-15.
- Bento J, Monteiro F, Sargento L, Vizcaino J, Monteiro J, Pilar Azevedo P, et al. Isolated ventricular noncompaction: a case report. Cases Journal. 2009;2:9312.
- Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36:493-500.
- Ritter M, Oechslin E, Sutsch G, Attenhofer C, Schneider J, Jenni R. Isolated Noncompaction of myocardium in adults. Mayo Clinic Proc. 1997;72:26-31.