|
Álvarez L Jiménez1, Alonso M Díez1, Martín A Blázquez1, Corral C Díez2, D Miminas3 Departments of 1General and Digestive Surgery and 2Pathology, Príncipe de Asturias University Hospital. Alcalá de Henares. Madrid, Spain; 3Department of Plastic and Reconstructive Surgery, Valles Hospital, Alcala de Henares. Madrid, Spain.
Corresponding Author:
Dr. Jiménez Álvarez L Email: laura.jimenezal@gmail.com
Abstract
Background: The solitary fibrous tumor (SFT) is a soft tissue tumor of fibroblastic origin. Most of the times, it develops in the thorax. Very infrequently, it appears in other locations such as the retroperitoneum or pelvis. Most cases have benign behavior, characterized by a slow localized growth, but 10-20% of cases are locally aggressive or metastasize in distant locations. Case Report: We present a case of extra-pleural SFT, in an unusual location, at the mesorectum. It reached 8 cm maximal diameter, and was accompanied by macroscopic traits of local malignancy. Histopathological examination and immunohistochemistry revealed a mesenchymal fusocellular/spindle cell tumor. Conclusion: Although an uncommon mesenchymal neoplasia, SFT should be kept in mind when making the differential diagnosis of pelvic masses. It should be considered as a neoplasm with a high risk of local recurrence or metastasis.
|
6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff241a2a000000ac06000001000e00 6go6ckt5b5idvals|952 6go6ckt5b5|2000F757Tab_Articles|Fulltext Introduction
The solitary fibrous tumor (SFT) is a soft tissue tumor of fibroblastic origin, composed of fusiform/spindle cells and possessing a great vascularization [ 1]; it is usually characterized by a slow localized growth [ 2]. Its incidence is very low, approximately 1.4 cases per million inhabitants. Most of the times, it develops in the thorax. Very infrequently, it appears in other locations such as the retroperitoneum or pelvis [ 3] with less than 100 cases described in these locations [ 4]. Most cases have benign behavior, but 10-20% of cases are locally aggressive or metastasize in distant locations [ 5]. Its diagnosis is the result of the combination of histopathology and immunohistochemistry, which makes it very difficult to diagnose at initial presentation [ 1]. We present a case of extra-pleural SFT, in an unusual location, at the mesorectum, where it reached a large size, and was accompanied by macroscopic and histopathological traits of local malignancy; it posed a diagnostic problem.
Case Report
A 65-year-old woman presented initially with leucorrhoea/vaginal discharge. Her past medical history included multiple previous abdominal surgery (right ureteral replacement with small intestine interposition following renal tuberculosis, hysterectomy with double adnexectomy for endometriosis) and allergy to iodine. A vaginal ultrasound unveiled a solid lesion of about 7 cm that seemed to relate to the rectum, so further study was initiated. A computed axial tomography (CT) of the abdomen and pelvis was performed [Fig.1] in which a mass of rounded morphology was identified, located in the right perirectal space, with well-defined margins, measuring 7.3×6.5×6.2 cm, heterogeneous, with hypodense lobulated areas, as well as peripheral calcifications. The lesion showed a fatty plane of separation from the rest of the pelvic structures without identifying a clear relation to any organ. It showed no infiltrative character. These findings suggested that it could be a sarcomatous type tumor. Colonoscopy revealed an extrinsic compression of the posterior surface of the upper rectum with intact rectal mucosa. The study was completed with a CT-guided needle aspiration biopsy, in which a proliferation of fusiform cells, without necrosis or frank cellular atypia was observed. Immunohistochemistry showed reactivity against vimentin, CD34, CD99 and Bcl2; it was negative against actin, desmin, S100, EMA, Factor XIIIa, HMB-45. These findings guided the initial working diagnosis of a neoplasia towards an extra-pleural SFT without ruling out mesenchymal neoplasia of greater aggressiveness.
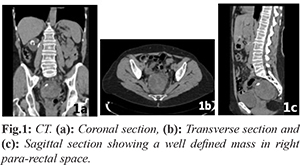
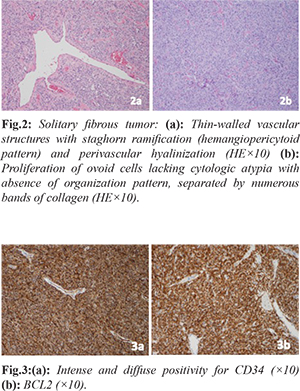
A laparotomy was performed in which a tumor was found occupying the whole of the pelvic space, apparently related to the mesorectum, poorly demarcated and without clear plane of separation from the adjacent structures. A low anterior rectal resection was performed, with block excision of the tumor along with the rectum and mesorectum, and mechanical colorectal anastomosis was performed at mid-rectum. Macroscopic examination showed an encapsulated nodular tumor of heterogeneous aspect of about 8 cm maximal diameter which seemed to originate from the mesorectum and did not appear to infiltrate the rectal wall. Microscopi-cally, it was a mesenchymal fusocellular/spindle cell tumor presenting a mitotic index of 4/10 HPF, with hyper-cellular areas, cytologic atypia, and intra-tumoral hemorrhage. Resection margins were affected [Fig.2]. Immunohistochemistry revealed expression of CD-34 and Bcl-2 [Fig.3] while CD117 was negative, pointing the diagnosis towards extra-pleural SFT with signs of malignancy. There was no evidence of lymphatic dissemination.
Post-operative period was uneventful. The patient was discharged from hospital on the sixth post-operative day. Given the existence of histological risk factors, the age of the patient and the size of the tumor, it was decided, in a Tumors Multidisciplinary Team meeting, to administer adjuvant radiotherapy, a total dose of 54 Gy with fractionation of 2Gy per session (5 sessions/week). So far, two follow up CT scans have been performed, at 2 and 6 months after surgery, without evidence suggesting relapse.
Discussion
The solitary fibrous tumor is a rare type of mesenchymal neoplasia whose most common location is the visceral pleura [ 6], although it has been reported in many other locations, such as skin, meninges, abdominal wall, retro-peritoneum and pelvis. It is estimated that approximately 34% of SFTs arise in the abdomino-pelvic region. However, among these, origin from the mesorectum is very rare, since there are only five cases reported in the literature [ 7]. Pelvic localization, according to most series studied, is associated with tumors having a larger size at the time of diagnosis than those at other locations, and a greater tendency for local recurrence [ 8]. In general, all SFTs are asymptomatic tumors that are usually diagnosed incidentally when performing an imaging test for another reason. As far as clinical presentation, it is usually related to the size of the tumor, its location and the compression of local structures [ 2] although associated paraneoplastic syndromes have also been described, such as hypertrophic osteoarthropathy and hypoglycemia [ 9]. Imaging tests have a fundamental role in diagnosis. Initial test is usually a computed axial tomography (CT) in which SFTs, regardless of location, are visualized as rounded, well demarcated and vascularized masses. Peripheral vascularization is frequent while calcifications are rare and may also present varying degrees of central necrosis. In magnetic resonance imaging (MRI) the lesion has the same characteristics as in CT, but in addition, up to 89% of the cases of SFT are hyper-intense in T2 [ 4]. In the pre-operative study, fine needle aspiration (FNA) and core biopsy (Tru-Cut) are also used, but generally with poor diagnostic performance. In FNA, numerous spindle and ovoid cells with homogeneous nuclei are often seen in a highly collagenous background, distributed in a disorganized way. However, that same image can be seen in many neoplasms, so to differentiate them, immunohistochemistry should be used. Nevertheless, the representativeness is very low, and its findings are not conclusive [10]. Differential diagnosis includes sarcomas, metastases or gastrointestinal stroma tumors (GISTs) [ 1]. Characteristically the tumor cells are positive for antibodies CD34, CD99 and vimentin, being negative for cytokeratin, smooth muscle actin, EMA and desmin in most cases [ 10]. In our case and following the CT findings, it was decided to perform a CT guided FNA of the lesion, obtaining satisfactory material for analysis. Immunohistochemistry of the sample revealed positivity of CD34, vimentin, CD99 and BCL-2, which pointed directly towards extra-pleural SFT as the working diagnosis. Given that, to obtain a definitive diagnosis, it was necessary to extract the complete tumor/specimen, we decided not to perform an MRI as it would not provide more information. Surgery is considered key to treatment [ 11], since resectability is the most important factor for long-term results. Complete excision, with microscopically free resection margins, is the factor that provides the best prognosis without local recurrences or distal metastases, even if the tumor has malignant characteristics [ 3]. Despite this, and especially for the pelvic SFTs, they present specific limitations for their complete excision, due to the anatomical characteristics of the pelvis [ 12]. Most of the series agree that extra-thoracic localization, size greater than 10 cm, affected resection margins, high rate of mitosis or presence of hemorrhage or intra-tumoral necrosis are related to a higher rate of local recurrence or the development of distant metastasis even many years after surgery [ 7, 13, 14]. Because of this, and given the unpredictability of their behavior, most authors recommend a very long post-operative follow-up [ 1, 7, 15] without existing clinical guidelines that determine either the frequency or the tests to be performed in this follow-up. Various adjuvant treatment options have been used. With regards to chemotherapy, different schemes have been used, such as intra-arterial chemoembolization with cisplatin and carboplatin or systemic agents such as imatinib mesylate in the unresectable SFTs and sunitib malate without any of them providing a clear improvement in long-term survival [ 1]. Adjuvant radiotherapy may provide a benefit in the case of incomplete resection and when risk factors for local recurrence are present, however, without evidence of impact on survival [ 3]. In our case, since the margins of resection were affected and the tumor presented other risk factors for local recurrence (pelvic localization, large size, high mitotic index, cellular atypia and intratumoral hemorrhage) it was decided to administer adjuvant radiotherapy. Conclusion
Although an uncommon mesenchymal neoplasia, SFT should be kept in mind when making the differential diagnosis of pelvic/intra-abdominal masses, since complete surgical resection is the most important prognostic factor. Despite its benign behavior, it should be considered as a neoplasm with a high risk of local recurrence or distant metastasis, so long-term follow-up is necessary.
Contributors: ALJ collected all the data, wrote the text and reviewed bibliography. AMD. MAB and DM attended and intervened the patient. All of them participated in the drafting and revision of the text. CCD made the histological analysis and provided critical inputs into the manuscript. ALJ will act as a study guarantor. All authors approved the final version of this manuscript. Funding: None; Competing interests: None stated.
References - Troja A, El-Sourani N, Antolovic D, Raab HR. Transsphinteric tumor resection in case of a pararectal solitary fibrous tumor. Int J Surg Case Rep. 2016; 19:159-162.
- Fibla JJ, Gómez G, Salord N, Penagos JC, Estrada G, León C. Tumor fibroso solitario pleural gigante. Cir Esp. 2005;77(5):290-292.
- Ishihara H, Omae K, Izuka J, Kobayashi H, Fukuda I, Kondo T, et al. Late recurrence of a malignant hypoglycemia-inducing pelvic solitary fibrous tumor secreting high-molecular-weight insulin-like growth factor-II: A case report with protein analysis. Oncology letters. 2016;12:479-484.
- Fernandez A, Conrad M, Gill RM, Choi WT, Kumar V, Behr S. Solitary fibrous tumor in the abdomen and pelvis: A case series with radiological findings and treatment recommendations. Clin Imaging. 2017;48:48-54.
- Beecham Chick JF, Chauhan NR, Madan R. Solitary fibrous tumors of the thorax: Nomenclature, epidemiology, radiologic and pathologic findings, differential diagnoses, and management. AJR 2013;200:W238-W248.
- Sanchez-Mora N, Cebollero-Presmanes M, Monroy V, Carretero-Albiñana L, Herranz-Aladro M, Álvarez-Fernández E. Tumor fibroso solitario pleural: características clinicopatologicas de una serie de casos y revisión de la bibliografía. Arch Bronconeumol. 2006; 42(2):96-99.
- Kawamura J, Tani M, Kida Y, Sumida K, Ogawa R, Kawasoe J, et al. Successful laparoscopic treatment of a giant solitary fibrous tumor of the mesorectum: A case report and literature review. Asian J Endosc Surg. 2017;10(1):51-54.
- Kawamura S, Nakamura T, Oya T. Advanced malignant solitary fibrous tumor in pelvis responding to radiation therapy. Pathol Int. 2007;57:213-218.
- Tricona-Garrón AB, Alfaro-Méndez G, Farrera-Torija J. Medina-Olivera E. Tumor fibroso solitario, reporte de un caso con presentación inusual. Rev Mex Urol. 2015;75(4):227-230.
- Ferreira B, Edgar J, Díaz P, Julio A. Tumor fibroso solitario de la pleura. Rev. Chilena de Cirugía. 2008;60:5.
- Cardillo G, Lococo F, Carleo F, Martelli M. Solitary fibrous tumors of the pleura. Curr Opin Pulm Med. 2012;18:339.
- Baldi GG, Stacchiotti S, Mauro V, Dei Tos AP, Gronchi A, Pastorino U, et al. Solitary fibrous tumor of all sites: Outcome of late recurrences in 14 patients. Clin Sarcoma Res. 2013;3:4.
- Hasegawa T, Matsuno Y, Shimoda T, et al. Extrathoracic solitary ?brous tumors: Their histological variability and potentially aggressive behavior. Hum Pathol. 1999; 30:1464-1473.
- Ailbhe C. O'Neill, Sree Harsha Tirumani, Woo S. Do, Abhishek R. Keraliya, Jason L. Hornick, Atul B. Shinagare, et al. Metastatic patterns of solitary fibrous tumors: A single-institution experience. American Journal of Roentgenology. 2017;2:1.
- Sánchez-Mora N, Cebollero-Presmanes M, Monroy V, Carretero-Albiñana L, Herranz-Aladro M, Álvarez-Fernández E. Tumor fibroso solitario: características clinicopatológicas de una serie de casos y revisión de la bibliografía. Arch. Bronconeumol. 2006;42(2):96-99.
|