6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff281a2a000000a906000001000c00
6go6ckt5b5idvals|953
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Pineoblastoma (PB) are rare brain tumors considered as a variant of supra-tentorial primitive neuroectodermal tumor. They are poorly differentiated, infiltrative and have significant potential for leptomeningeal and extra-cranial dissemination [
1]. Pineoblastoma have predilection for the pediatric population, where as in adults it accounts for approximately less than 1% of primary central nervous system tumors [
2]. Due to rarity of pineoblastoma in adults, optimal treatment strategies and prognosis are yet to be determined. Hence we report our experience in the management of pineoblastoma in an adult patient.
Case Report
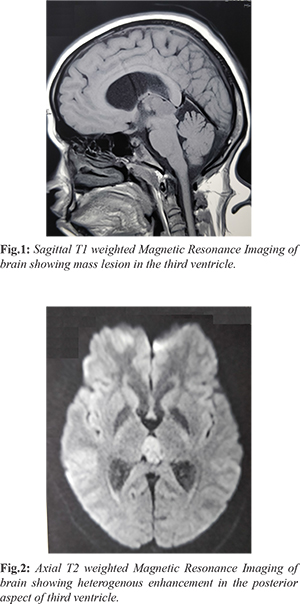
A 27 year old female was initially evaluated elsewhere for the complaints of double vision associated with headache for one month duration. Clinically she had no focal neurological deficits. An opthalmologic examination revealed no evidence of papilledema and field of vision were normal. Magnetic Resonance Imaging (MRI) brain showed a heterogenous enhancing polypoidal mass lesion in the posterior aspect of third ventricle measuring 1.9×1.5 cm in axial plane and 1.6 cm craniocaudally with mild ventriculomegaly of bilateral ventricle [Fig.1,2]. She underwent peritrocular craniotomy, supra-cerebellar infra-tentorial approach and gross total excision of lesion. Her post-operative period was uneventful. Histopathological examination showed fragments of neuroepithelial tumor of variable cellularity, perivascular pseudo-rosettes and Homer-Wright rosettes. Few fragments were hypo-cellular with fibrous stroma and tumor cells forming pineocytomatous rosettes. The features were consistent with pineoblastoma, WHO grade IV.
Immunohistochemistry (IHC) done showed the tumor cells were strongly and diffusely positive for synaptophysin. Glial fibrillary acidic protein (GFAP) and epithelial membrane antigen (EMA) were negative. Patient received post-operative radiotherapy comprising craniospinal irradiation at a dose of 36 Gy in 20 fractions followed by a local 18 Gy boost to the tumor site for a total dose of 54 Gy with concurrent chemotherapy with weekly vincristine.
Discussion
Pineal region tumors encompass a heterogenous group of neoplasms consisting of germ cell tumors, glial tumors, pineal parenchymal tumors and other miscellaneous tumors and cysts. Pineal parenchymal tumors (PPTs) originate from the cells of pineal gland called pinealocytes and represents only 0.3-1% of all primary central nervous system tumors [
3]. The current World Health Organization classification of PPTs includes pineocytoma (Grade I), PPTs of intermediate differentiation (Grade II or III), papillary tumor of the pineal region (Grade II or III) and pineoblastoma (Grade IV) [
4]. PB are more common in children and in adults it accounts for less than 10% of published cases. Although pineoblastoma may occur in young to middle-aged adults, a 2012 literature review by Tate et al. concluded that pediatric and adult pineoblastomas behave differently and should therefore be separately considered when analyzing their response to surgical and adjuvant therapy [
5]. The clinical features of pineoblastoma are related to the mass effect in the region that includes headache, nausea, double vision, abnormalities in eye movements. In our case the patient presented with double vision associated with headache.
Radiographically there will be areas of calcification spread throughout the lesion (i.e, “exploded calcifications”), a feature of pineal parenchymal tumors. Calcifications are best seen on computed tomography (CT), but can be visible on MRI. Diffusion-weighted MRI shows restricted diffusion in pineoblastomas, similar to those seen in other intracranial high-grade neoplasms that correspond with increased cellularity [
6]. On MRI, PB are usually characterized by hypo-intensity to iso-intensity on T1 weighted images and iso-intensity to hyper-intensity on T2 weighted images.
Histopathologically PB are less cellular composed of patternless sheet of densely packed mitiotically active small cells with round to irregular nuclei and scant cytoplasm, necrosis is common. They can have Homer-Wright rosettes. In our case histopathological examination showed fragment of neuroepithelial tumor of variable cellularity, perivascular pseudorosettes and Homer-Wright rosettes. Few fragments were hypocellular with fibrous stroma and tumor cells forming pineocytomatous rosettes. Immunohistochemistry (IHC) done showed tumor cells were strongly and diffusely positive for synaptophysin favouring PB.
The appropriate treatment for PB have not been determined as the incidence rate is extremely low, particularly in adults and only a few described cases with limited follow up and outcome studies are available [
7]. The recommended treatment for adults with PB includes maximum surgical resection followed by adjuvant craniospinal radiotherapy and a boost to the entire posterior fossa, with or without the addition of chemotherapy [
8]. Tumors originating from the pineal region have varied propensities for recurrence and patterns of dissemination. A few described cases reported that radiotherapy aided in controlling the tumor and improving survival [
9]. Systemic chemotherapy includes several different protocols that vary widely which includes combination of following drugs cisplatin or carboplatin, cyclophosphamide drugs. In our case the patient underwent surgery, radiotherapy and chemotherapy. Patient did not develop any major complications during the course of treatment and currently she is on follow up.
Conclusion
The pineoblastoma have significant potential for leptomeningeal and extra-cranial dissemination; adjuvant craniospinal radiotherapy aids in controlling the tumor and improving survival.
Contributors: RB collected all the data, wrote the text, reviewed bibliography and was involved in patient management. MN, AR attended and intervened the patient. All of them participated in the drafting and revision of the text. MN will act as a study guarantor. All authors approved the final version of this manuscript.
Funding: None; Competing interests: None stated.
References
- Luther N, Stetler WR Jr, Dunkel IJ, Christos PJ, Wellons JC 3rd, Souweidane MM. Subarachnoid dissemination of intraventricular tumors following simultaneous endoscopic biopsy and third ventriculostomy. J Neurosurg Pediatr. 2010;5:61-67.
- Lee JY, Wakabayashi T, Yoshida J. Management and survival of pineoblastoma:an analysis of 34 adults from the brain tumor registry of Japan. Neurol Med Chir (Tokyo). 2005;45(3):132-141.
- Cuccia V, Rodriguez F, Palma F, Zuccaro G. Pineoblastomas in children. Childs Nerv Syst. 2006;22(6):577-585.
- Tumours of the pineal region. In: WHO Classification of Tumours of the Central Nervous System, 3rd, Louis D, Ohgaki H, Wiestler O, Cavenee WK (eds), WHO Publication Center, Albany, NY 2007.
- Tata M, Sughrue ME, Rutkowski MJ, Kane AJ, Aranda D, McClinton L, et al. The long-term postsurgical prognosis of patients with pineoblastoma. Cancer. 2012;118(1):173-179.
- Choudhri AF, Whitehead MT, Siddiqui A, Klimo P Jr, Boop FA. Diffusion characteristics of pediatric pineal tumors. Neuroradiol J. 2015;28(2):209-216.
- DeBoer R, Batjer H, Marymont M, Goldmann S, Wlker M, Gottardi-Littell N, et al. Response of an adult patient with pineoblastoma to vorinostat and retinoic acid. J Neurooncol. 2009;95:289-292.
- Preafico F, Massimino M, Gandola L, Cefalo G, Mazza E, Landonio G, et al. Survival of adults treated with medulloblastoma using Pediatric Protocols. Eur J Cancer. 2005;41(9):1304-1310.
- Hinkes BG, Von Hoff K, Deinlein F, Warmuth-Metz M, Soerensen N, Timmermann B. Childhood pineoblastoma; experiences from the prospective multicentre trials HIT-SKK87, HIT-SKK92 and HIT 91. J Neuro Oncol. 2007;81(2):217-223.