6go6ckt5b8|3000F7576AC3|Tab_Articles|Fulltext|0xf1ff24912b0000003805000001000900
6go6ckt5b5idvals|968
6go6ckt5b5|2000F757Tab_Articles|Fulltext
Introduction
Davison (1954) first introduced the term pallidopyramidal syndrome (PPS). He described a series of five patients who presented with the triad of progressive parkinsonism, spasticity and dystonia. By now about 18 cases have been reported in English literature [
1]. Most of these cases are familial with an autosomal recessive mode of inheritance and presented in young adulthood. These patients were initially responsive to levodopa, but later developed levodopa induced dyskinesias and loss of response to levodopa. Here we are presenting a rare case of sporadic pallidopyramidal syndrome with some interesting additional clinical features resembling Kufor Rakeb syndrome. To the best of our knowledge this is the first case of a sporadic PPS with Kufor Rakeb like features being reported from India.
Case Report
A 28 years old right handed Hindu male, farmer by occupation, presented with 4 years history of insidious onset progressive slowness of daily activities and gait difficulty with frequent falls for last 3 years. He also had developed rest tremors in hands, drooling of saliva, change in speech and urinary incontinence followed by cognitive decline for the last 2 years. He was born of a nonconsanguineous marriage and had a normal birth history and developmental milestones. He was a high school graduate, married and father of a child.
Four years ago the patient gradually started developing difficulty in walking, his speed of walking reduced and he started walking with stiff legs taking short steps. He generally slowed down in carrying out all his routine activities. His problems gradually worsened and he developed rest tremors of right hand which also involved the left hand over the next few months. Hand tremors were prominent at rest and during voluntary activity and diminished during sleep. Progressively he became incapable of carrying out activities related to farming and household chores. About a year later, he also developed a change in speech. He became progressively dysarthric with low volume, indistinct and barely understandable speech accompanied with drooling of saliva. Gradually the family members noticed change in his personality and behavior. He became more and more apathetic, dull and poorly interactive with occasional anger outbursts. He gradually became incontinent for urine and feces without awareness or any social concern. He also became progressively forgetful for recent events although he could remember past events clearly. However, he did not forget familiar paths, had no difficulty recognizing and naming things and people. There was no history of headache, seizures, hallucinations, self mutilating or violent behavior. There were no complaints of any visual or hearing disturbances or any sensory symptoms. There was no history of exposure to drugs, toxins, insecticides or organophosphates or jaundice, blood loss, head trauma or sexual promiscuity. There was no history of a similar illness in any family member.
The initial examination revealed a regular pulse of 74/minute. Blood pressure was 114/80 mmHg in supine position with no postural drop. He was conscious, alert and cooperative. Hindi MMSE score was 15/29. On higher mental function examination his orientation to time, place and person, attention and vigilance were impaired. Registration and immediate recall were normal but recent memory was impaired. Remote memory was intact. He showed impairment on tests for visual and verbal memory as well. Tests for frontal lobe function also revealed impairment. Praxis, insight and judgment were normal. His speech was slow, hypophonic, effortless with preserved repetition, naming and auditory comprehension and his facies were masked and had continuous drooling of saliva. On cranial nerve examination visual acuity and fundus examination were normal. Horizontal eye movements were full range with slow saccades and saccadic pursuits but vertical gaze restriction was present both in upward and downward gaze, upgaze being more impaired than downgaze. There was no nystagmus but had frequent blepharospasm [Video 1].
Spasticity was present in both the lower limbs (grade 4 modified Ashworth spasticity scale) and cogwheel rigidity in both hands. Power was normal in all the muscle groups. All deep tendon reflexes were brisk and bilateral plantar reflexes were extensor. He could sit with support with a flexed posture and had significant postural instability. There were perioral tremors, resting pill rolling tremors in hands (grade 3 UPDRS scale) with dystonic posturing, facial–faucial–finger minimyo clonus, oculo-gyric dystonic spasm and striatal toes [Video 2]. The UPDRS score was 149/206=72.3% and Schwaband England activities of daily living scale ADL was 10%. Sensory system examination was normal. There were no intention tremors, coordination in lower limbs could not be tested due to spasticity. He walked with a stooped posture and had a spastic gait with frequent freezing and marked postural instability (UPDRS Grade 3).
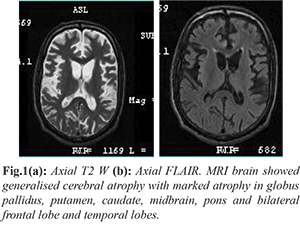
This patient clinically was a PPS with early parkinsonian features, dystonia and spasticity, which manifested at 24 years of age without any family history and consanguinity. We considered the differential diagnosis of Wilson’s disease, young onset parkinson’s disease [PARK-9 (Kufor Rakeb), PARK-14, PARK 15], neuro-degeneration with brain iron accumulation (NBIA), aceruloplasminemia, juvenile Huntington disease and adult onset Gaucher’s disease. MRI brain showed generalised cerebral atrophy with marked atrophy in globus pallidus, putamen, caudate, midbrain and pons and bilateral frontal lobe and temporal lobes [Fig.1].
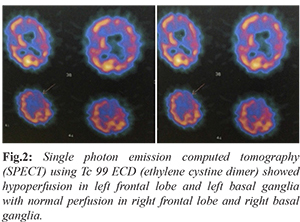
Single photon emission computed tomography (SPECT) using Tc 99 ECD (ethylene cystine dimer) showed hypoperfusion in left frontal lobe and left basal ganglia with normal perfusion in right frontal lobe and right basal ganglia [Fig.2]. Routine blood work up including complete blood count, renal and liver function, blood sugar, HbA1c, serum electrolytes, thyroid function, vitamin B12, ESR, ANA, HIV, HBsAg, anti HCV, serum calcium and parathormone were normal. KF ring was absent. 24 hour urinary copper (52 microgram/day) and serum ceruloplasmin (35 mg/dL) values were within normal limits. No acanthocytes were seen on a freshly prepared thick peripheral blood smear. Ultrasonography of the abdomen and radiographs of the chest, hip and pelvis were normal. Huntingtin gene test for CAG repeats was negative. Nerve conduction study and electroencephalography were normal. Wilson’s disease was excluded based on the absence of KF ring, normal 24 hr urinary copper and serum ceruloplasmin. Normal fundus and HbA1C excluded aceruloplasminemia. Absence of iron deposition or eye of tiger in MRI brain was against NBIA as a diagnosis. CAG repeat for Huntingtin gene was negative. Absence of hypersplenism, hepatosplenomegaly and bony lesions made the possibility of Gaucher’s disease less likely. SPECT study revealed hypoperfusion in left frontal lobe and left basal ganglia. Genetic studies could not be done due to financial constraints.
He was prescribed L-dopa/carbidopa 125 mg four times daily and trihexyphenydil 2 mg thrice daily. He responded well with significant functional improvement in speech, gait, resting tremors and apathy. After initiation of syndopa he showed marked improvement on UPDRS scale 85/206= 41.3% (before levodopa-149/206= 72.3%) and Schwab and England activities of daily living scale ADL=40% (before levodopa ADL=10%). After few days, he developed levodopa induced dyskinesias and intention tremors, which were not present before levodopa therapy. Interestingly after taking levodopa he developed intention tremors in both hands, which might be due to unmasking of mild cerebellar dysfunction. He was discharged on L-dopa/carbidopa 125 mg thrice daily and trihexyphenydil 2 mg thrice daily.
Discussion
The first ever case of young parkinsonism was reported in 1919, which was followed in 1922 by Hallervorden Spatz reporting a family of five sisters with brownish discoloration of globus pallidus which we know as the present day neurodegeneration with brain iron accumulation (NBIA). Davison in 1954 first reported five patients with progressive parkinsonism, spasticity and dystonia who had pallidal and pyramidal degeneration with blue brown discoloration of globus pallidus and introduced the clinical term pallidopyramidal degeneration [
1].
Pallidopyramidal syndromes (PPS) are an uncommon group of parkinsonian disorders with additional features like pyramidal signs, dystonia, cognitive impairment and rarely cerebellar involvement; without evidence suggestive of other disease entities like Wilson’s disease or multiple system atrophy which may present with a combination of extra-pyramidal, pyramidal, cerebellar or cognitive features. Only 18 cases have so far been reported in literature of PPS and most of them are of an autosomal recessive inheritance. We report this rare case of pallido pyramidal syndrome of sporadic variety with additional clinical features resembling Kufor Rakeb syndrome [
1,
2].
Our patient had a PPS with additional features similar to Kufor Rakeb syndrome. Kufor Rakeb syndrome was originally reported by Aldin et al. in a consanguineous couple [
1]. It is a variety of young onset Parkinson’s disease with autosomal recessive mode of transmission (PARK 9), occurring due to homozygous or compound heterozygous mutation in the ATP13A2 gene, which encodes a lysosomal type 5 ATPase, on chromosome 1p36. The usual age of onset is between 11 -16 yrs, however, recently many cases have been reported with age of onset in 4th and 5th decades [
4]. These patients presented with early parkinsonian features, spasticity, hyperreflexia, extensor plantar, mild cerebellar dysfunction, supranuclear gaze palsy, dementia, facial-finger–faucial minimyoclonus, blepharospasm and oculo-gyric dystonic spasm [
2]. Such patients are initially responsive to levodopa, but later they become less responsive to levodopa along with appearance of drug induced dyskinesias.
Our patient had a PPS with additional features and simulated the clinical profile of young onset Parkinson disease resembling the Kufor Rakeb variety (PARK 9). Parkinsonian features in our patient were low volume monotonous speech, mask like facies, freezing of gait, flexed posture, pill rolling resting tremors, cogwheel rigidity in both hands and postural instability. Additional characteristic features were vertical gaze palsy (upward > downward), blepharospasm, facial-faucial–finger minimyoclonus, oculogyric dystonic spasms, dystonia, impaired recent memory, verbal and visual memory, apathy, spasticity (clasp knife type), hyper-reflexia, bilateral extensor plantars. He also showed a significant clinical improvement with levodopa induced dyskinesias. Interestingly, he manifested another rare feature of appearance of intention tremors after dopaminergic therapy, suggesting unmasked mild cerebellar dysfunction.
Our patient neither had a positive family history nor was a product of consanguinity. There was no question of a false paternity also. All the reported cases of Kufor Rakeb (PARK 9) are of autosomal recessive inheritance while our case is a sporadic variety, very rarely reported in literature. Our case could therefore be classified clinically as a sporadic pallido pyramidal syndrome with additional features resembling Kufor Rakeb. It may be attributable to a new mutation or probably PPS is a heterogeneous condition [
5,
6].
Conclusion
It has been argued that cases presenting with Davison’s triad might not represent a distinct clinicopathological group and might belong to a group of young onset “nigral” parkinsonisms (rather than “pallidal” parkinsonism described in the classical PPS) or dopa responsive dystonias. Davison’s cases could also have been representatives of the modern neurodegeneration with brain iron accumulation which encompasses clinical entities presenting with dystonia parkinsonism (PPS triad) and evidence of iron accumulation on imaging. NBIAs have been further classified into subtypes with the identification of causative genes (PANK2, PLA2G6, FTL, ATP 13A2). Eleanna Kara et al. have argued recently in a review published in 2013 that NBIA classifies clinical entities (Davison’s PPS triad) based on iron accumulation and genetic loci, but iron accumulation is not consistently found in all these disorders and the complete genetic landscape of these disorders is far from understood. They propose that rather than the term NBIA, the disorders presenting with the clinical Davison’s triad should be reclassified as PPS with or without iron accumulation on MRI. This would allow for a simpler clinical and pathological classification of these syndromes and would allow further incorporation of the genetic discoveries. Therefore, PPS with or without iron accumulation seems to be a clinically relevant and useful entity [
1].
With the recent discovery of the genetic mutations associated with many parkinsonian pyramidal syndromes many of these syndromes would eventually be attributable to particular genetic loci. But the knowledge of the genetic loci responsible for such disorders is far from complete. From a clinical standpoint it is important to classify these disorders based on clinical syndromes which may guide further pathologic and genetic studies in similarly presenting cases.
We describe this rare case of sporadic pallidopyramidal syndrome with additional clinical features resembling Kufor Rakeb syndrome, without evidence of brain iron accumulation on imaging. To the best of our knowledge it is the first report of such a case from India.
Contributors: BS, PD: manuscript writing, literature review, patient management; AP, RP: critical inputs into the manuscript and patient management. PD will act as a study guarantor. All authors approved the final version of this manuscript and are responsible for all aspects of this study.
Funding: None; Competing interests: None stated.
References
- Kara E, Hardy J, Houlden H. The pallidopyramidal syndromes: nosology, aetiology and pathogenesis. Curr Opin Neurol. 2013;26(4):381-394.
- Paisán-Ruiz C1, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacs in mutations. Mov Disord. 2010;25(12):1791-800.
- Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A, Wriekat AL. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet. 2001;38:680-682.
- Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T, et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord. 2010;25:1929-1937.
- Kalita J, Misra UK, Das BK. Sporadic variety of pallido-pyramidal syndrome. Neurol India. 2003;51:383-384.
- Panagariya A, Sharma B, Dev A. Pallido-pyramidal syndrome: A rare entity. Indian J Med Sci. 2007;61:156-157.